laporan ske b blok 22 kelompok 7
DESCRIPTION
laporan kelompokTRANSCRIPT

1
LAPORAN TUTORIAL
SKENARIO B BLOK 22
Disusun Oleh: KELOMPOK 7
Avyandara Janurizka 04121401013
Muhammad Ramzie 04121401019
M. Alniroman Y. 04121401025
Reijefki Irlastua Simbolon 04121401032
Nur Annisa Faradina 04121401034
Intan Fajrin Karimah 04121401046
Achmad Randi Raharjo 04121401051
Vivi Miliarti 04121401061
Adisti Meirizka 04121401070
M. Yufimar Riza Fadilah 04121401076
Abdur Rozak 04121401080
M. Fakhri Altyan 04121401082
Tutor : dr. Fifie Julianita, SpPA
FAKULTAS KEDOKTERAN
UNIVERSITAS SRIWIJAYA

2
KATA PENGANTAR
Puji syukur kami haturkan kepada Allah SWT atas segala rahmat dan karunia-Nya
sehingga kami dapat menyelesaikan laporan tutorial yang berjudul “Laporan Tutorial Skenario
B Blok 22” sebagai tugas kompetensi kelompok. Salawat beriring salam selalu tercurah kepada
junjungan kita, nabi besar Muhammad SAW beserta para keluarga, sahabat, dan pengikut-
pengikutnya sampai akhir zaman.
Kami menyadari bahwa laporan tutorial ini jauh dari sempurna. Oleh karena itu kami
mengharapkan kritik dan saran yang bersifat membangun guna perbaikan di masa mendatang.
Dalam penyelesaian laporan tutorial ini, kami banyak mendapat bantuan, bimbingan dan saran.
Pada kesempatan ini, kami ingin menyampaikan syukur, hormat, dan terimakasih kepada :
1. Allah SWT, yang telah merahmati kami dengan kelancaran diskusi tutorial,
2. dr. Fifie Julianita, SpPA selaku tutor kelompok 7
3. teman-teman sejawat FK Unsri,
4. semua pihak yang telah membantu kami.
Semoga Allah SWT memberikan balasan pahala atas segala amal yang diberikan kepada
semua orang yang telah mendukung kami dan semoga laporan tutorial ini bermanfaat bagi kita
dan perkembangan ilmu pengetahuan. Semoga kita selalu dalam lindungan Allah SWT. Amin.
Palembang, 10 Desember 2014
Kelompok 7

3
DAFTAR ISI
Judul…………………………………………………………………………………1
Kata Pengantar………………………………………………………………………2
Daftar Isi…………………………………………………………………………….3
Kegiatan tutorial ............................................................................................................4
I. Skenario
II. Klarifikasi Istilah…………………………………………………......................5
III. Identifikasi Masalah……………………….…………………………………...6
IV. Analisis Masalah……………………….……………………….……………....7
V. Hipotesis……………………………...………………………...……………….15
VI. Template……….……………………..…………………………………………15
VII. Learning issue........................................................................................................27
VIII. Kerangka Konsep……………………..………………………………………55
IX. Kesimpulan………………………………………………………..……………55
Daftar Pustaka……………………………………………………...……..........56

4
KEGIATAN TUTORIAL
Tutor : dr. Fifie Julianita, SpPA
Moderator : M. Alniroman Y
Sekretaris Meja 1 : Abdur rozak
Sekretaris Meja 2 : Adisti Meirizka
Pelaksanaan : 15 Desember 2014 dan 17 Desember 2014
13.00 WIB- 15.00 WIB
Peraturan selama tutorial :
1. Angkat tangan sebelum berbicara
2. Dilarang makan dan minum
3. Penggunaan gadget hanya untuk keperluan diskusi tutorial

5
I. Skenario
A 9 years old girl came to the Moh. Hoesin Hospital with complain of pale and
abdominal distention. She lives in Kayu Agung. She has been already hospitalizied
two times before (2009 and 2010) in Kayu Agung General Hospital and always got
blood transfusion. Her younger brother, 7 years, looks taller than her. Her uncle was
died when he was 21 years olddue to the similar disease like her.
Physical examination:
Compos mentis, anemia (+), wide epicantus, prominent upper-jaw
Hr: 94x/mnt, rr: 27x, td: 100/70 mmhg, temp: 36,7
Heart and lung: withn normal limit
Abdomen: hepatic enlargement ¼ x 1/4 , spleen: schoeffner iii
Extremities: pallor palm of hand. Others: normal
Laboratory result:
Hb: 7,6 gr/dl, ret: 1,8%, wbc: 10,2 x109, thrombocyte: 267x109/lt, diff. Count:
0/2/0/70/22/6
Blodd film: anisocytosis, poikylocytosis, hypochrome, target cell (+)
Mcv: 64 (fl), mch: 21 (pg), mchc: 33 (gr/dl), si within normal limit, tibc within
normal limit, serum ferritin: within normal limit
II. Klarifikasi istilah
a. Pale : pucat
b. Abdominal distention : proses peningkatan tekanan abdomen yang menghasilkan
peningkatan tekanan dalam perut dan menekan diding perut
c. Blood transfusion : pemasukkan darah lengkap/komponen darah secara
langsung ke dalam aliran darah
d. Anemia : berkurangnya jumlah eritrosit atau kadar hemoglobin
eritrosit yang kurang dari normal
e. Epicantus : lipatan kulit vertical pada kedua sisi hidung yang kadang-
kadang menutupi kantus sebelah dalam

6
f. Prominent upper-jaw :penonjolan rahang atas
g. Schoeffner iii :pembesaran lien telah ditemukan 1 cm diatas umbilicus
h. Pallor : pucat
i. Anisocytosis : adanya eritrosit dalam darah yang menunjukkan variasi
ukuran yang besar
j. Poikylocytosis : adanya eritrosit dengan keragaman bentuk yang abnormal
didalam darah
k. Hypochrome : penurunan abnormal kandungan hemoglobin dalam
eritrosit
l. Target cell : sel yang pipih dengan diameter yang besar dan dapat
terlihat pada thalassemia, penyakit obstruktif dan penyakit sel sabit, dll
m. SI : kandunganbesi yang terdapatdalam serum
n. Tibc : kemampuan total transferrin untukmengikatbesi
o. Serum ferritin : kompleks besi apoferrtin yang merupakan bentuk utama
penyimpanan besi dalam tubuh
III. Identifikasi masalah
a. A 9 years old girl came to the moh. Hoesinhosppital with complain of pale and
abdominal distention.
b. She lives in Kayu Agung. She has been already hospitalizied two times before
(2009 and 2010) in Kayu Agung General Hospital and always got blood
transfusion.
c. Her younger brother, 7 years, looks taller than her. Her uncle was died when he
was 21 years olddue to the similar disease like her.
d. Physical examination:
Compos mentis, anemia (+), wide epicantus, prominent upper-jaw

7
Hr: 94x/mnt, rr: 27x, td: 100/70 mmhg, temp: 36,7
Heart and lung: withn normal limit
Abdomen: hepatic enlargement ¼ x 1/4 , spleen: schoeffner iii
Extremities: pallor palm of hand. Others: normal
e. Laboratory results:
Hb: 7,6 gr/dl, ret: 1,8%, wbc: 10,2 x109, thrombocyte: 267x109/lt, diff. Count:
0/2/0/70/22/6
Blodd film: anisocytosis, poikylocytosis, hypochrome, target cell (+)
Mcv: 64 (fl), mch: 21 (pg), mchc: 33 (gr/dl), si within normal limit, tibc within
normal limit, serum ferritin: within normal limit
IV. Analisis masalah
a. A 9 years old girl came to the moh. Hoesinhosppital with complain of pale and
abdominal distention.
i. Apakah hubungan usia, jenis kelamin dengan penyakit ?
Jawab:
Pada thalassemia mayor, gejala klinis dapat terlihat pada usia
dibawah 1 tahun. Penyakit thalassemia diturunkan secara autosomal
resesif. Sehingga tidak berpengaruh terhadap jenis kelamin.
ii. Apakah etiologi dan mekanisme dari pale dan abdominal distention ?
Jawab:
Warna merah dari darah manusia disebabkan oleh hemoglobin
yang terdapat didalam sel darah merah. Hemoglobin terdiri atas zat besi
dan protein yang dibentuk oleh rantai globin alpha dan rantai globin beta.
Pada penderita thalasemia beta , produksi rantai globin beta tidak ada atau
berkurang, sehingga hemoglobin yang dibentuk berkurang. Selain itu
berkurangnya rantai globin beta mengakibatkan rantai globin alpha
berlebihan dan akan saling mengikat membetnuk suatu benda yang

8
menyebabkan sel darah merah mudah rusak. Berkurangnya produksi
hemoglobin dan mudah rusaknya sel darah merah mengakibatkan
penderita pucat atau anemia dan kadar hemoglobinnya rendah.
Mekan i sme
Ke l a inan gene t i k (de l e s i pada gen yang mengkode
p ro t e in g lob in d i kromosom 11 atau 16) → Tidak terbentuknya salah
satu atau kedua rantai globin (Rantai β tidak terbentuk) →
peningkatan relative rantai α → rantai α berikatan dengan rantai γ
membentuk HbF (α2γ2) → peningkatan HbF → m e n g e n d a p d i
m e m b r a n ( H e i n z b o d i e s ) → R B C m u d a h dihancurkan →
Penurunan jumlah hemoglobin → (oksigenasi ke perifer berkurang)
→ pucat.
Abdominal distention :
Disitensi abdomen terjadi karena adanya penumpukan cairan ,
udara atau karena ada massa dan organomegali pada rongga abdomen.
Pada penderita thalasemia , distensi abdomen terjadi karena pembesaran
hati dan limpa. Limpa berfungsi membersihkan sel darah yang rusak. Pada
penderita thalasemia , sel darah merah yang rusak sangat berlebihan
sehingga kerja limpa sangat berat. Akibatnya limpa membengkak, selain
itu tugas limpa lebih diperberat untuk memproduksi sel darah merah lebih
banyak
Mekanisme
Ke la inan gene t i k (de l e s i pada gen yang mengkode
p ro t e in g lob in d i kromosom 11 atau 16) → Tidak terbentuknya salah
satu atau kedua rantai globin (Rantai β tidak terbentuk) →
peningkatan relative rantai α → rantai α berikatan dengan rantai γ
membentuk HbF (α2γ2) → peningkatan HbF → m e n g e n d a p d i
m e m b r a n ( H e i n z b o d i e s ) → R B C m u d a h d i h a n c u r k a n

9
( d i h a t i , l i m p a , d a n s i s t e m r e t i k u l o e n d o t e l i a l l a i n )
→ Pen ingka t an ke r j a ha t i dan l impa → Hepatosplenomegali
→ Distensi abdomen.
b. She lives in kayuagung. She has been already hospitalizied two times before
(2009 and 2010) in kayuagung general hospital and always got blood transfusion.
i. Apa hubungan tempat tinggal dengan penyakit yang diderita ?
Jawab:
Pada daerah endemi malaria, prevalensi thalasemianya sangat
tinggi karena penderita thalasemia resisten terhadap malaria. Sumatera
Selatan termasuk daerah yang sangat tinggi prevalensi thalasemia,
sehingga resiko A yang tinggal di Kayu Agung untuk menderita
thalasemia tinggi.
ii. Apa makna dari masuk rumah sakit dan transfusi dua kali ?
Jawab:
Pada penderita thalassemia dengan transfusi regular merupakan
kriteria dari thalassemia mayor, aktivitas berlebih akan menimbulkan
kelelahan. Oleh karena itu diperlukan transfusi darah rutin agar kebutuhan
tubuh kembali tercukupi. Jika tidak maka anemia dapat berulang.
iii. Apa indikasi dari transfusi darah ?
Jawab:
1. Kehilangan darah akut, bila 20–30% total volume darah hilang dan
perdarahan masih terus terjadi.
2. Anemia berat
3. Syok septik (jika cairan IV tidak mampu mengatasi gangguan sirkulasi
darah dan sebagai tambahan dari pemberian antibiotik)
4. Memberikan plasma dan trombosit sebagai tambahan faktor
pembekuan, karena komponen darah spesifik yang lain tidak ada

10
5. Transfusi tukar pada neonatus dengan ikterus berat.
6. Hb dibawah 7 g/dl dan menunjukkan gejala hemodinamik
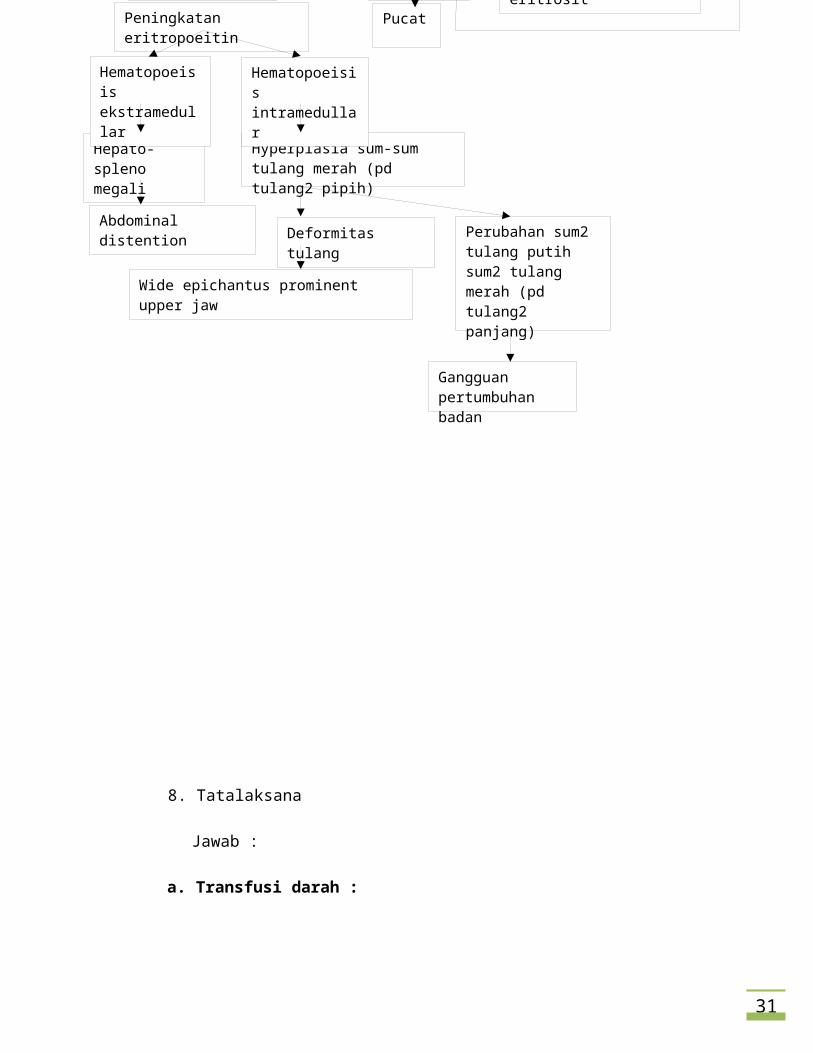
c. Her younger brother, 7 years, looks taller than her. Her uncle was died when he
was 21 years olddue to the similar disease like her.
i. Apa hubungan perbedaan tinggi badan antara adiknya dengan dia ?
Jawab:
1. Pada pasien thalasemia, terjadi destruksi dini eritrosit sehingga
sumsum tulang merah berkompensasi dengan cara meningkatkan
eritropoiesis. Sumsum tulang merah terdapat di tulang pipih seperti os
maxilla, os frontal, dan os parietal. Hal ini mengakibatkan tulang-
tulang tersebut mengalami penonjolan dan pelebaran. Namun,
destruksi dini sel darah merah terus berlanjut sehingga sumsum tulang
putih yang normalnya berfungsi untuk membangun bentuk tubuh dan
pertumbuhan berubah fungsi menjadi sumsum tulang merah yang
menghasilkan eritrosit. Sumsum tulang putih terdapat pada tulang-
tulang panjang seperti os tibia, os fibula, os femur, os radius, dan os
ulna. Perubahan fungsi tulang-tulang ini dari pembangun tubuh
menjadi pembentuk eritrosit mengakibatkan terhambatnya
pertumbuhan A.
2. Massa jaringan eritropetik yang membesar tetapi inefektif bisa
menghabiskan nutrient sehingga menyebabkan retardasi pertumbuhan
3. Penimbunan besi pada pasien thalassemia dapat merusak organ
endokrin sehingga terjadi kegagalan pertumbuhan dan gangguan
pubertas.
4. terjadi gangguan tumbuh kembang yang kemungkinan terjadi akibat
kurangnya oksigen dan nutrisi pada jaringan

11
ii. Apa hubungan riiwayat keluarga dengan penyakit ?
Jawab:
Thalasemia merupakan suatu kelainan genetik yang diturunkan,
yaitu merupakan suatu penyakit autosomal resesif dengan delesi
dikromosom 11 (Thalasemia beta) atau 16 (Thalasemia alpha) sehingga
kemungkinan paman A juga menderita thalasemia.
Gejala pada A cocok dengan gejala thalasemia betha mayor yang
dapat mematikan bila tidak ditangani dengan benar (diberi transfusi darah
secara rutin, atau dilakukan transplantasi sumsum tulang ). Dalam kasus
thalasemia mayor, kematian terjadi pada dekade kedua atau ketiga,
biasanya akibat gagal jantung kongestif atau aritmia jantung
d. Physical examination:
Compos mentis, anemia (+), wide epicantus, prominent upper-jaw
Hr: 94x/mnt, rr: 27x, td: 100/70 mmhg, temp: 36,7
Heart and lung: withn normal limit
Abdomen: hepatic enlargement ¼ x 1/4 , spleen: schoeffner iii
Extremities: pallor palm of hand. Others: normal
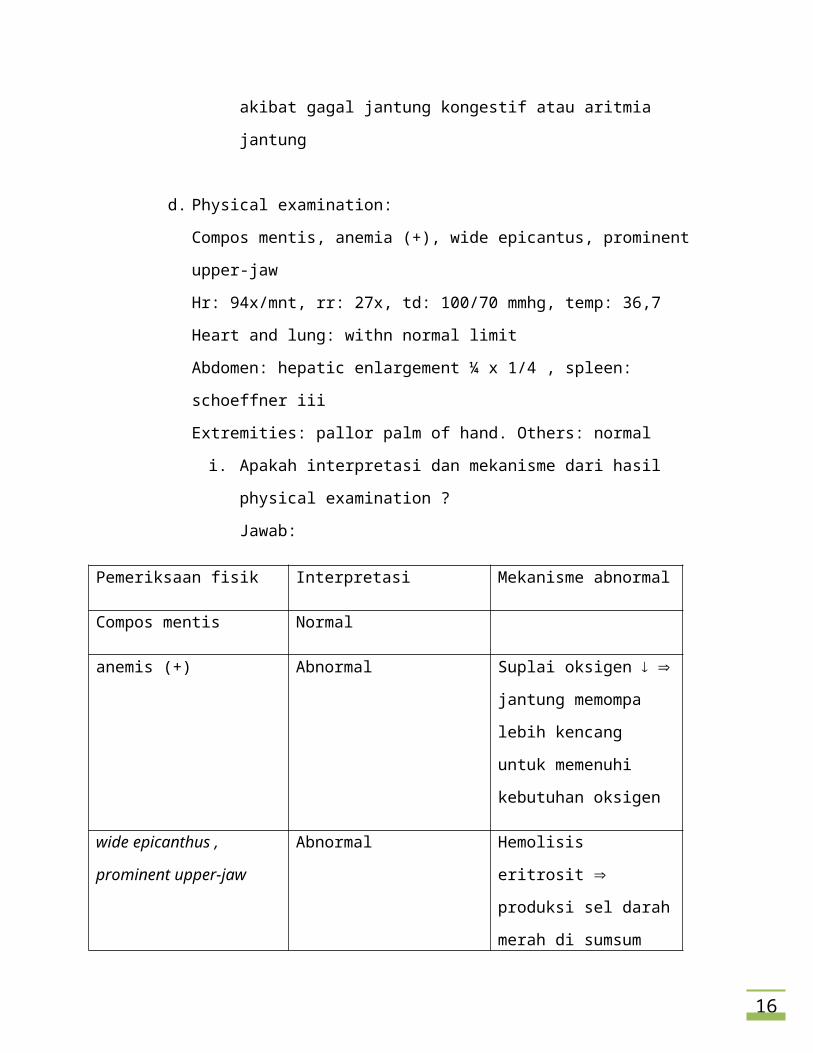
i. Apakah interpretasi dan mekanisme dari hasil physical examination ?
Jawab:
Pemeriksaan fisik Interpretasi Mekanisme abnormal
Compos mentis Normal
anemis (+) Abnormal Suplai oksigen jantung
memompa lebih kencang
untuk memenuhi
kebutuhan oksigen
wide epicanthus , prominent
upper-jaw
Abnormal Hemolisis eritrosit
produksi sel darah merah di
sumsum tulang

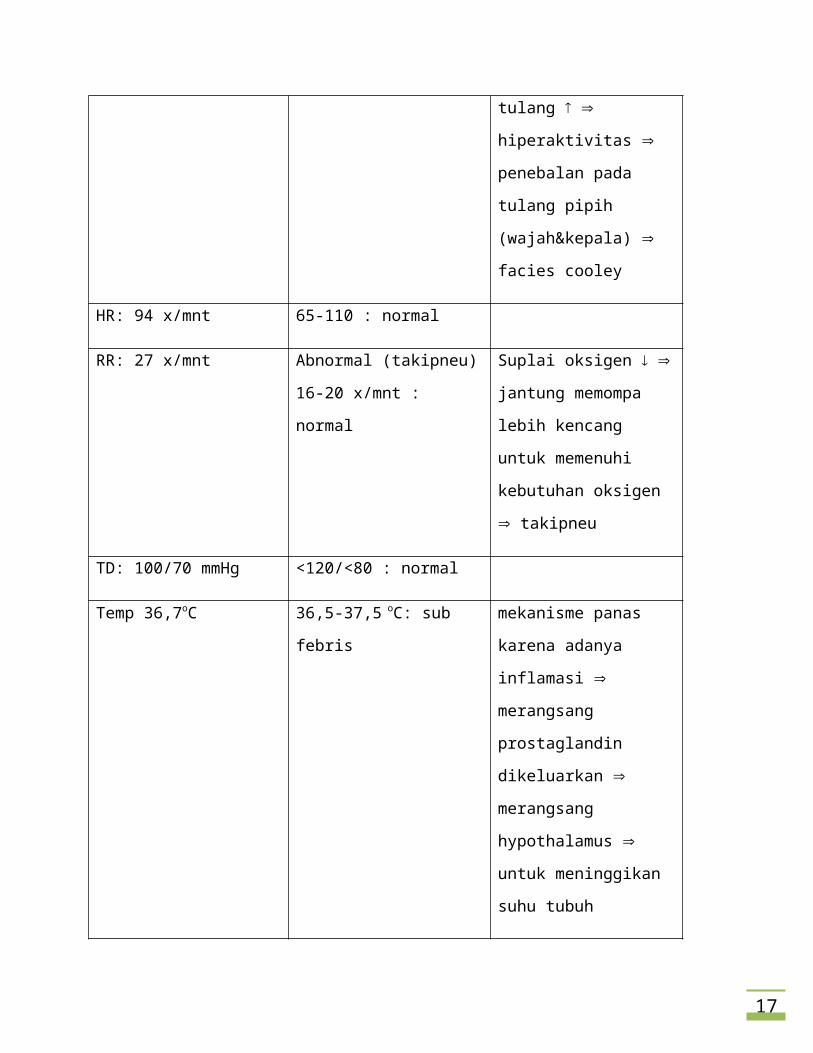
12
hiperaktivitas penebalan
pada tulang pipih
(wajah&kepala) facies
cooley
HR: 94 x/mnt 65-110 : normal
RR: 27 x/mnt Abnormal (takipneu)
16-20 x/mnt : normal
Suplai oksigen jantung
memompa lebih kencang
untuk memenuhi
kebutuhan oksigen
takipneu
TD: 100/70 mmHg <120/<80 : normal
Temp 36,7oC 36,5-37,5 oC: sub febris mekanisme panas karena
adanya inflamasi
merangsang prostaglandin
dikeluarkan merangsang
hypothalamus untuk
meninggikan suhu tubuh
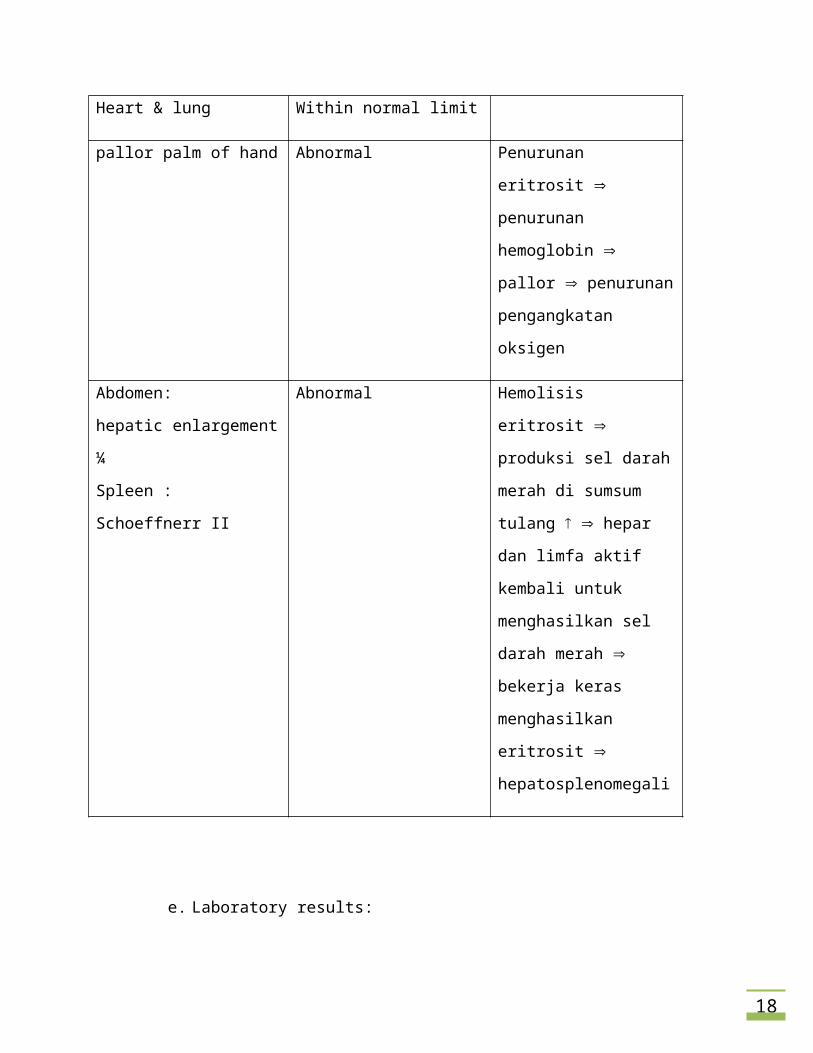
Heart & lung Within normal limit
pallor palm of hand Abnormal Penurunan eritrosit
penurunan hemoglobin
pallor penurunan
pengangkatan oksigen
Abdomen:
hepatic enlargement ¼
Spleen : Schoeffnerr II
Abnormal Hemolisis eritrosit
produksi sel darah merah di
sumsum tulang hepar
dan limfa aktif kembali
untuk menghasilkan sel
darah merah bekerja

13
keras menghasilkan
eritrosit
hepatosplenomegali
e. Laboratory results:
Hb: 7,6 gr/dl, ret: 1,8%, wbc: 10,2 x109, thrombocyte: 267x109/lt, diff. Count:
0/2/0/70/22/6
Blodd film: anisocytosis, poikylocytosis, hypochrome, target cell (+)
Mcv: 64 (fl), mch: 21 (pg), mchc: 33 (gr/dl), si within normal limit, tibc within
normal limit, serum ferritin: within normal limit
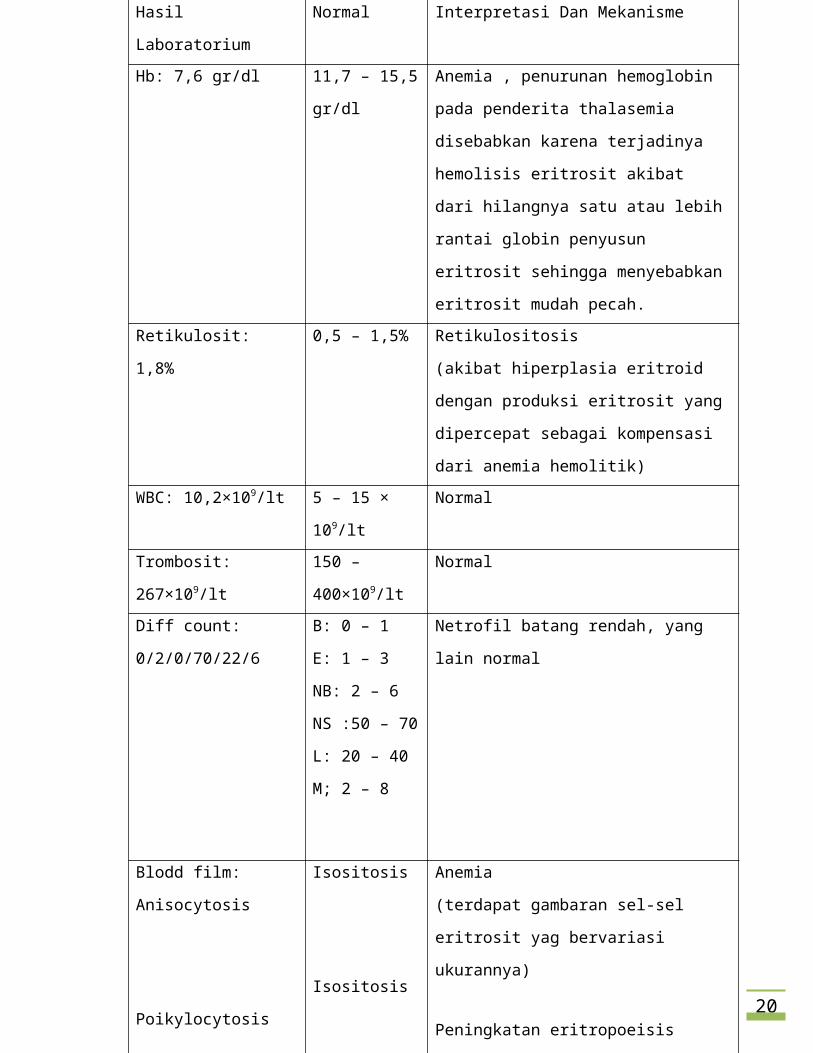
i. Apakah interpretasi dan mekanisme dari hasil laboratory results ?
Jawab:

14
Hasil Laboratorium Normal Interpretasi Dan Mekanisme
Hb: 7,6 gr/dl 11,7 – 15,5 gr/dl Anemia , penurunan hemoglobin pada
penderita thalasemia disebabkan karena
terjadinya hemolisis eritrosit akibat dari
hilangnya satu atau lebih rantai globin
penyusun eritrosit sehingga menyebabkan
eritrosit mudah pecah.
Retikulosit: 1,8% 0,5 – 1,5% Retikulositosis
(akibat hiperplasia eritroid dengan produksi
eritrosit yang dipercepat sebagai kompensasi
dari anemia hemolitik)
WBC: 10,2×109/lt 5 – 15 × 109/lt Normal
Trombosit: 267×109/lt 150 – 400×109/lt Normal
Diff count: 0/2/0/70/22/6 B: 0 – 1
E: 1 – 3
NB: 2 – 6
NS :50 – 70
L: 20 – 40
M; 2 – 8
Netrofil batang rendah, yang lain normal
Blodd film: Anisocytosis
Poikylocytosis
Hypochrome
Target cell (+)
Isositosis
Isositosis
Normokrom
-
Anemia
(terdapat gambaran sel-sel eritrosit yag
bervariasi ukurannya)
Peningkatan eritropoeisis (gambaran sel-sel
eritrosit dengan bentuk yang beragam)
Rendahnya Hb dalam darah (warna pucat
pada bagian tengah eritrosit yang lebih besar
dari biasanya)
↑ resistensi osmotik membran eritrosit
(adanya peningkatan eritropoeisis tetapi tidak
efektif sehingga menghasilkan sel-sel eritrosit

15
V. Hipotesis
Anak perempuan berumur 9 tahun diduga mengalami anemia hemolitik et causa
thalassemia
VI. Template
1. How to diagnose
Jawab :
1. Anamnesis
Keluhan utama : timbul karena anemia yaitu pucat, gangguan nafsu makan, gangguan tumbuh
kembang dan perut membesar karena pembesaran lien dan hati. Pada umumnya keluhan ini
mulai timbul pada usia 6 bulan Keluhan.
tempat tinggal di daerah Endemik Thalassemia-β
Riwayat keluhan : ada salah satu atau lebih keluarga yang juga menderita penyakit yang sama,
Riwayat pucat yang berlangsung kronis. Pernah / sering menerima transfusi darah, mudah
terkena infeksi.
2. Pemeriksaan fisis
a. Pucat
b. Bentuk muka mongoloid (facies Cooley)
c. Dapat ditemukan ikterus
d. Gangguan pertumbuhan
e. Splenomegali dan hepatomegali yang menyebabkan perut membesar
3. Pemeriksaan penunjang
a. Darah tepi
Hb rendah dapat sampai 2-3 g%
Gambaran morfologi eritrosit : mikrositik hipokromik, sel target, anisositosis berat
dengan makroovalositosis, mikrosferosit, polikromasi, basophilic stippling, benda
Howell-Jolly, poikilositosis dan sel target. Gambaran ini lebih kurang khas.
Retikulosit meningkat.
b. Sumsum tulang (tidak menentukan diagnosis)
16
Hiperplasi sistem eritropoesis dengan normoblas terbanyak dari jenis asidofil.
Granula Fe (dengan pengecatan Prussian biru) meningkat.
c. Pemeriksaan khusus :
Hb F meningkat : 20%-90% Hb total
Elektroforesis Hb : hemoglobinopati lain dan mengukur kadar Hb F.
Pemeriksaan pedigree: kedua orangtua pasien thalassemia mayor merupakan trait
(carrier) dengan Hb A2 meningkat (> 3,5% dari Hb total).
4. Pemeriksaan lain
Foto Ro tulang kepala;
Gambaran hair on end, korteks menipis, diploe melebar dengan trabekula tegak lurus
pada korteks.
Foto tulang pipih dan ujung tulang panjang;
Perluasan sumsum tulang sehingga trabekula tampak jelas.
5. Diagnosis banding
Thalasemia minor :
Anemia kurang besi
Anemia karena infeksi menahun
Anemia pada keracunan timah hitam (Pb)
Anemia sideroblastik
2. Dd
Jawab :
Berdasarkan pemeriksaan subjektif dan objektif, serta insidensi/epidemik penyakit di daerah
tempat tinggal pasien (Kayu Agung), diperoleh diagnosis banding sebagai berikut:
Thalassemia Malaria Anemia Defisiensi Besi
Pucat + + +
Distensi Abdomen + + -
Transfusi Darah + - -
17
Anemis + - +
Hepatosplenomegaly + + -
Hb < + + +
Retikulosit > + -
Anisoytosis + +
Poikylositosis + +
Hipokrom + + +
Target sel + - +/-
MCV < + + +
MCH normal + + +
MCHC < + + +
Serum iron normal + + -
TIBC normal + + -
Serum ferritin normal + + -
3. Wd
Jawab :
anemia hemolitik e.c thalasemia
4. Pemeriksaan penunjang
Jawab :
Pemeriksaan laboratorium yang perlu untuk menegakkan diagnosis thalasemia ialah
a. Darah
Pemeriksaan darah yang dilakukan pada pasien yang dicurigai menderita thalasemia adalah
1. Darah rutin
Kadar hemoglobin menurun. Dapat ditemukan peningkatan jumlah lekosit, ditemukan
pula peningkatan dari sel PMN. Bila terjadi hipersplenisme akan terjadi penurunan
dari jumlah trombosit.
2. Hitung retikulosit
Hitung retikulosit meningkat antara 2-8 %.
18
3. Gambaran darah tepi
Anemia pada thalassemia mayor mempunyai sifat mikrositik hipokrom. Pada
gambaran sediaan darah tepi akan ditemukan retikulosit, poikilositosis, tear drops sel
dan target sel.
4. Serum Iron & Total Iron Binding Capacity
Kedua pemeriksaan ini dilakukan untuk menyingkirkan kemungkinan anemia terjadi
karena defisiensi besi. Pada anemia defisiensi besi SI akan menurun, sedangkan TIBC
akan meningkat.
5. LFT
Kadar unconjugated bilirubin akan meningkat sampai 2-4 mg%. bila angka tersebut
sudah terlampaui maka harus dipikir adanya kemungkinan hepatitis, obstruksi batu
empedu dan cholangitis. Serum SGOT dan SGPT akan meningkat dan menandakan
adanya kerusakan hepar. Akibat dari kerusakan ini akan berakibat juga terjadi
kelainan dalam faktor pembekuan darah.
b. Elektroforesis Hb
Diagnosis definitif ditegakkan dengan pemeriksaan eleltroforesis hemoglobin. Pemeriksaan
ini tidak hanya ditujukan pada penderita thalassemia saja, namun juga pada orang tua, dan
saudara sekandung jika ada. Pemeriksaan ini untuk melihat jenis hemoglobin dan kadar Hb
A2. petunjuk adanya thalassemia α adalah ditemukannya Hb Barts dan Hb H. Pada
thalassemia β kadar Hb F bervariasi antara 10-90%, sedangkan dalam keadaan normal
kadarnya tidak melebihi 1%.
c. Pemeriksaan sumsum tulang
Pada sumsum tulang akan tampak suatu proses eritropoesis yang sangat aktif sekali. Ratio
rata-rata antara myeloid dan eritroid adalah 0,8. pada keadaan normal biasanya nilai
perbandingannya 10 : 3.
d. Pemeriksaan roentgen
Ada hubungan erat antara metabolisme tulang dan eritropoesis. Bila tidak mendapat tranfusi
dijumpai osteopeni, resorbsi tulang meningkat, mineralisasi berkurang, dan dapat diperbaiki
dengan pemberian tranfusi darah secara berkala. Apabila tranfusi tidak optimal terjadi

19
ekspansi rongga sumsum dan penipisan dari korteknya. Trabekulasi memberi gambaran
mozaik pada tulang. Tulang terngkorak memberikan gambaran yang khas, disebut dengan
“hair on end” yaitu menyerupai rambut berdiri potongan pendek pada anak besar.
5. Epidemiologi
Jawab :
Dilihat dari distribusi geografiknya maka thalasemia β banyak dijumpai dii Mediterania,
timur Tengah, India/Pakistan, dan Asia Tenggara. Di Siprus dan Yunani lebih banyak
dijumpai varian β+, sedangkan di Asia Tenggara lebih banyak varian βo. Jika diilukiskan
dalam peta dunia, seolah-olah membentuk sebuah sabuk thalasemia, dimana Indonesia
termasuk didalamnya. Sedangkan thalasemia α sering di jumpai di Asia Tenggara.
6. Etiologi
Jawab :
Kelainan genetik : dalam hal kurangnya satu atau lebih atau tidak terbentuknya rantai globin (α atau β) dari Hb.• Mutasi gen β globin pada kromosom 11 yang mengkode rantai β• Delesi gen α globin pada kromosom 16 yang mengkode rantai α
7. Pathogenesis
Jawab :
Hemoglobin dewasa atau HbA mengandung dua rantai αdan dua rantai . Ditandai oleh dua
gen globin yang bertempat pada masing-masing dari dua kromosom nomor 11. Dan, dua
pasang gen α-globin yang fungsional berada pada setiap kromosom nomor 16. Struktur dasar gen
α-globin dan, begitu juga langkah-langkah yang terlibat dalam biosintesis rantai globin adalah
sama. Setiap gen globin memiliki tiga rangkaian pengkodean (ekson) yang diganggu oleh dua
rangkaina peratara (intron). Pengapitan sisi 5’ gen globin merupakan serentetan “rangkaian
20
promoter” yang tidak dapat diterjemahkan, yang diperlukan untuk inisiasi sintesis mRNA -
globin.
Seperti pada semua gen eukariotik, biosintesis rantai globin mulai dengan transkripsi gen
globin di dalam nucleus. Transkripsi mRNA awal mengandung suatu salinan seluruh gen,
termasuk semua ekson dan intron. Precursor mRNA yang besar ini mengalami beberapa
modifikasi pascatranskripsi (proses) sebelum diubah menjadi mRNA sitoplasma dewasa yang
siap untuk translasi yaitu penyambungan dua intron dan mengikat kembali ekson. mRNa dewasa
yang terbentuk meninggalkan nucleus dan menjadi terkait ribosom pada tempat translasi berlaku.
Jalur ekspresi gen α-globin sangat serupa. (Buku Ajar Patologi II, Robbins & Kumar –
Jakarta :EGC, 1995).
Thalassemia diartikan sebagai sekumpulan gangguan genetik yang mengakibatkan
berkurang atau tidak ada sama sekali sintesis satu atau lebih rantai globin (Weatherall and Clegg,
1981). Abnormalitas dapat terjadi pada setiap gen yang menyandi sintesis rantai polipeptid
globin, tetapi yang mempunyai arti klinis hanya gen-βdan gen-α. Karena ada 2 pasang gen-α,
maka dalam pewarisannya akan terjadi kombinasi gen yang sangat bervariasi. Bila terdapat
kelainan pada keempat gen-α maka akan timbul manifestasi klinis dan masalah. Adanya kelainan
gen-α lebih kompleks dibandingan dengan kelainan gen-β yang hanya terdapat satu pasang.
Gangguan pada sintesis rantai-α dikenal dengan penyakit thalassemia-α, sedangkan gangguan
pada sintesis rantai-β disebut thalassemia-β.
Kelainan klinis pada sintesis rantai globin-alfa dan beta dapat terjadi, sebagai berikut:
a. Silent carrier yang hanya mengalami kerusakan 1 gen, sehingga pada kasus ini tidak
terjadi kelainan hematologis. Identifikasi hanya dapat dilakukan dengan analisis
molekular menggunakan RFLP atau sekuensing.
b. Bila terjadi kerusakan pada 2 gen-α atau thalassemia-α minor atau carrier thalassemia-
α menyebabkan kelainan hematologis.
c. Bila terjadi kerusakan 3 gen-α yaitu pada penyakit HbH secara klinis termasuk
thalassemia intermedia.

21
Mutasi / delesi gen globin β
Hemoglobin menurun
Sintesis rantai β globin menurun atau terhenti
Peningkatan rantai globin α yg tidak berpasangan
Berikatan dng rantai γ globin
HbF meningkat
Afinitas thd oksigen meningkat
Hipoksia jaringan
Presipitasi rantai α pd eritroblas di sumsum tulang
Eritropoeisis tidak efektif
ANEMIA
Presipitasi rantai α pd membrane eritrosit
Fleksibilitas eritrosit ↓
↑ hemolisis di limpa
Pemendekan umur eritrosit
d. Pada Hb-Bart’s hydrop fetalis disebabkan oleh kerusakan keempat gen globin-alfa dan
bayi terlahir sebagai Hb-Bart’s hydrop fetalis akan mengalami oedema dan asites
karena penumpukan cairan dalam jaringan fetus akibat anemia berat.
e. Pada thalassemia-β mayor bentuk homozigot (β0) dan thalassemia-β minor (β+)
bentuk heterozigot yang tidak menunjukkan gejala klinis yang berat.
Gangguan yang terjadi pada sintesis rantai globin-α ataupun-β jika terjadi pada satu atau
dua gen saja tidak menimbulkan masalah yang serius hanya sebatas pengemban sifat (trait atau
carrier). Thalassemia trait disebut uga thalassemia minor tidak menunjukkan gejala klinis yang
berarti sama halnya seperti orang normal kalaupun ada hanya berupa anemia ringan. Kadar Hb
normal aki-laki: 13,5 – 17,5 g/dl dan pada wanita: 12 – 14 g/dl. Namun demikian nilai indeks
hematologis, yaitu nilai MCV dan MCH berada di bawah nilai rentang normal. Rentang normal
MCV: 80 – 100 g/dl, MCH: 27 – 34 g/dl

22
8. Tatalaksana
Jawab :
a. Transfusi darah :
Hb penderita dipertahankan antara 8 g/dl sampai 9,5 g/dl. Dengan kedaan ini akan
memberikan supresi sumsum tulang yang adekuat, menurunkan tingkat akumulasi
besi, dan dapat mempertahankan pertumbuhan dan perkembangan penderita.
Pemberian darah dalam bentuk PRC (packed red cell), 3 ml/kg BB untuk setiap
kenaikan Hb 1 g/dl.
b. Medikamentosa
1) Vitamin E 200-400 IU setiap hari sebagai antioksidan dapat memperpanjang
umur sel darah merah.

23
2) Asam folat 2-5 mg/hari untuk memenuhi kebutuhan yang meningkat.
3) Vitamin C 100-250 mg/hari selama pemberian kelasi besi, untuk meningkatkan
efek kelasi besi.
4) Bila kadar ferritin serum atau serum iron meningkat:
Pemberian iron chelating agent (desferoxamine): diberikan setelah kadar feritin
serum sudah mencapai 1000 mg/l atau saturasi transferin lebih 50%, atau sekitar
10-20 kali transfusi darah.Desferoxamine, dosis 25-50 mg/kg berat badan/hari
subkutan melalui pompa infus dalam waktu 8-12 jam dengan minimal selama 5
hari berturut setiap selesai transfusi darah. Atau desferopron oral.
c. Bedah
Splenektomi merupakan prosedur pembedahan utama yang digunakan pada pasien
dengan thalassemia. Limpa diketahui mengandung sejumlah besar besi nontoksik (yaitu,
fungsi penyimpanan). Limpa juga meningkatkan perusakan sel darah merah dan
distribusi besi. Fakta-fakta ini harus selalu dipertimbangkan sebelum memutuskan
melakukan splenektomi.. Limpa berfungsi sebagai penyimpanan untuk besi nontoksik,
sehingga melindungi seluruh tubuh dari besi tersebut. Pengangkatan limpa yang terlalu
dini dapat membahayakan.
Sebaliknya, splenektomi dibenarkan apabila limpa menjadi hiperaktif,
menyebabkan penghancuran sel darah merah yang berlebihan dan dengan demikian
meningkatkan kebutuhan transfusi darah, menghasilkan lebih banyak akumulasi besi.
Imunisasi pada penderita ini dengan vaksin hepatitis B, vaksin H.Influenzae tipe B, dan
vaksin polisakarida pneumokokus diharapkan, dan terapi profilaksis penisilin juga
dianjutkan.
Splenektomi, dengan indikasi:
Anak usia >6 tahun
Limpa yang terlalu besar, sehingga membatasi gerak penderita, menimbulkan
peningkatan tekanan intraabdominal dan bahaya terjadinya ruptur. Hipersplenisme
ditandai dengan peningkatan kebutuhan transfusi darah atau kebutuhan suspensi
eritrosit (PRC) melebihi 250 ml/kg berat badan dalam 1 tahun.
d. Transplantasi sumsum tulang (TST)

24
Pengobatan thalassemia β yang berat dengan transplantasi sumsum tulang allogenik pertama kali
dilaporkan lebih dari satu dekade yang lalu, sebagai alternatif dari pelaksanaan klinis standar dan
saat ini diterima dalam pengobatan thalassemia β.
9. Pencegahan
Jawab :
Kelahiran penderita thalassemia dapat dicegah dengan 2 cara. Pertama adalah mencegah
perkawinan antara 2 orang pembawa sifat thalassemia. Kedua adalah memeriksa janin
yang dikandung oleh pasangan pembawa sifat, dan menghentikan kehamilan bila janin
dinyatakan sebagai penderita thalassemia (mendapat kedua gen thalassemia dari ayah dan
ibunya).VSebaiknya semua orang Indonesia dalam masa usia subur diperiksa
kemungkinan membawa sifat thalassemia beta. Karena frekuensi pembawa sifat
thalassemia beta di Indonesia berkisar antara 6-10%, artinya setiap 100 orang ada 6
sampai 10 orang pembawa sifat thalassemia beta. Terlebih lagi apabila ada riwayat
seperti di bawah ini, pemeriksaan pembawa sifat thalassemia sangat dianjurkan: Ada
saudara sedarah yang menderita thalassemia beta.Kadar hemoglobin relatif rendah antara
10-12 g/dl, walaupun sudah minum obat penambah darah seperti zat besi. Ukuran sel
darah merah lebih kecil dari normal walaupun keadaan Hb normal.
Diagnosis prenatal melalui beberapa tahap. Tahap pertama adalah pemeriksaan
ibu janin yang meliputi pemeriksaan darah tepi lengkap dan analisis hemoglobin. Bila ibu
dinyatakan pembawa sifat thalassemia beta maka pemeriksaan dilanjutkan ke tahap kedua
yaitu suami diperiksa darah tepi lengkap dan analisis hemoglobin. Bila suami juga
membawa sifat thalassemia maka suami-isteri ini diperiksa DNAnya untuk menentukan
jenis kelainann pada gen globin beta. Selanjutnya diambil jaringan janin (villi choriales
atau jaringan ari-ari) pada saat janin berumur 10-12 minggu untuk diperiksa DNAnya.
Bila janin ternyata hanya membawa satu belah gen globin beta yang mengalami kelainan
(gen thalassemia beta) atau sama sekali tidak membawa gen thalassemia beta maka
kehamilan dapat diteruskan dengan aman. Tetapi bila janin ternyata membawa kedua
belah gen thalassemia yang artinya janin akan menderita thalassemia beta maka
penghentian kehamilan dapat menjadi pilihan.

25
Pengambilan jaringan janin dari ari-ari dilakukan dengan menusukkan jarum
melalui jalan lahir atau dinding perut ke dalam alat kandungan clan menembus ke ari-ari,
kemudian pada daerah ari-ari yang disebut villi choriales diambil dengan cara aspirasi
sejumlah jaringan tersebut untuk bahan pemeriksaan DNA. Prosedur ini dilakukan oleh
dokter ahli kandungan yang sudah berpengalaman melakukan tindakan ini. Prosedur ini
dilakukan pada kehamilan 11 minggu. Tindakan ini mempunyai risiko keguguran sebesar
2-3%. Cara lain untuk mendapat sel dari janin adalah dengan pengambilan cairan amnion
yang baru dapat dilakukan pada kehamilan 15 minggu. Risiko abortus pada prosedur ini
adalah 1%.
10. Komplikasi
Jawab :
a. Hemosiderosis
b. Cardiac disease (akibat iron overload pada myocardium)
c. Kematian
d. Fraktur patologis
e. Disfungsi organ
Akibat anemia yang berat dan lama, sering terjadi gagal jantung. Transfusi darah yang
berulang-ulang dan proses hemolisis menyebabkan kadar besi dalam darah tinggi, sehingga
ditimbun dalam berbagai jaringan tubuh seperti hepar, limpa, ku.lit, jantung dan lainnya. Hal ini
dapat mengakibatkan gangguan fungsi alat tersebut. Limpa yang besar mudah rupture akibat
trauma yang ringan. Kadang-kadang thalasemia disertai oleh tanda hipersplenisme seperti
leukopenia dan trombopenia.
Kematian terutama disebabkan oleh infeksi dan gagal jantung.
Kelebihan Fe (khususnya pada pemberian transfusi)
Komplikasi pada jantung, contoh constrictive pericarditis to heart failure and arrhythmias.
Komplikasi pada hati, contoh hepatomegali sampai cirrhosis.
Komplikasi jangka panjang, contoh HCV.
Komplikasi hematologic, contoh VTE.

26
Komplikasi pada endokrin, seperti endokrinopati, DM.
Gagal tumbuh karena diversi dari sumber kalori untuk eritropoesis.
11. Prognosis
Jawab :
Quo Vitam: malam
Quo Fungsionam: malam
Prognosis thalassemia tergantung pada tipe dan derajat keparahan thalassemia. Perjalanan
klinis thalassemia sangat bervariasi mulai dari yang ringan atau terkadang asimptomatik
sampai keadaan yang berat dan mengancam jiwa.
Thalassemia beta homozigot umumnya meninggal pada usia muda dan jarang mencapai
usia dekade ke 3, walaupun digunakan antibiotik untuk mencegah infeksi dan pemberian
chelating agent untuk mengurangi hemosiderosis.
12. Kdu
Jawab :
3A
Mampu membuat diagnosis klinik berdasarkan pemeriksaan fisik dan pemeriksaan-
pemeriksaan tambahan yang diminta oleh dokter (misalnya:X-ray).Dokter dapat
memutuskan dan memberi terapi pendahuluan,serta merujuk ke spesialis yang
relevan(bukan kasus gawat darurat)
VII. Learning issue
1. Proses haematopoesis
Jawab :

27
- Hematopoiesis merupakan proses produksi (mengganti sel yang mati) dan perkembangan
sel darah dari sel induk / asal / stem sel, dimana terjadi proliferasi, maturasi dan
diferensiasi sel yang terjadi secara serentak.- Proliferasi sel menyebabkan peningkatan atau pelipat gandaan jumlah sel, dari satu sel
hematopoietik pluripotent menghasilkan sejumlah sel darah. Maturasi merupakan proses
pematangan sel darah, sedangkan diferensiasi menyebabkan beberapa sel darah yang
terbentuk memiliki sifat khusus yang berbeda-beda.- Tempat terjadinya hematopoiesis pada manusia :- 1. Embrio dan Fetus- a. Stadium Mesoblastik, Minggu ke 3-6 s/d 3-4 bulan kehamilan : Sel-sel mesenchym di
yolk sac. Minggu ke 6 kehamilan produksi menurundiganti organ-organ lain- b. Stadium Hepatik, Minggu ke 6 s/d 5-10 bulan kehamilan : Menurun dalam waktu
relatif singkat. Terjadi di Limpa, hati, kelenjar limfe- c. Stadium Mieloid, Bulan ke 6 kehamilan sampai dengan lahir, pembentukan di sumsum
tulang : Eritrosit, leukosit, megakariosit.- 2. Bayi sampai dengan dewasa- Hematopoiesis terjadi pada sumsum tulang, normal tidak diproduksi di hepar dan limpa,
keadaan abnormal dibantu organ lain.- a. Hematopoiesis Meduler (N)- Lahir sampai dengan 20 tahun : sel sel darah → sumsum tulang. Lebih dari 20 tahun :
corpus tulang panjang berangsur – angsur diganti oleh jaringan lemak karena produksi
menurun.- b. Hematopoiesis Ekstrameduler (AbN)- Dapat terjadi pada keadaan tertentu, misal: Eritroblastosis foetalis, An.Peniciosa,
Thallasemia, An.Sickle sel, Spherositosis herediter, Leukemia. Organ – organ
Ekstrameduler : Limpa, hati, kelenjar adrenal, tulang rawan, ginjal, dll - Macam – macam hematopoiesis- 1. Seri Eritrosit (Eritropoesis)

28
- Perkembangan eritrosit ditandai dengan penyusutan ukuran (makin tua makin kecil),
perubahan sitoplasma (dari basofilik makin tua acidofilik), perubahan inti yaitu nukleoli
makin hilang, ukuran sel makin kecil, kromatin makin padat dan tebal, warna inti gelap.- Tahapan perkembangan eritrosit yaitu sebagai berikut :- a. Proeritroblas- Proeritroblas merupakan sel yang paling awal dikenal dari seri eritrosit. Proeritroblas
adalah sel yang terbesar, dengan diameter sekitar 15-20µm. Inti mempunyai pola
kromatin yang seragam, yang lebih nyata dari pada pola kromatin hemositoblas, serta
satu atau dua anak inti yang mencolok dan sitoplasma bersifat basofil sedang. Setelah
mengalami sejumlah pembelahan mitosis, proeritroblas menjadi basofilik eritroblas.- b. Basofilik Eritroblas- Basofilik Eritroblas agak lebih kecil daripada proeritroblas, dan diameternya rata-rata
10µm. Intinya mempunyai heterokromatin padat dalam jala-jala kasar, dan anak inti
biasanya tidak jelas. Sitoplasmanya- yang jarang nampak basofil sekali.- c. Polikromatik Eritroblas (Rubrisit)- Polikromatik Eritoblas adalah Basofilik eritroblas yang membelah berkali-kali secara
mitotris, dan menghasilkan sel-sel yang memerlukan hemoglobin yang cukup untuk dapat
diperlihatkan di dalam sediaan yang diwarnai. Setelah pewarnaan Leishman atau Giemsa,
sitoplasma warnanya berbeda-beda, dari biru ungu sampai lila atau abu-abu karena
adanya hemoglobin terwarna merah muda yang berbeda-beda di dalam sitoplasma yang
basofil dari eritroblas. Inti Polikromatik Eritroblas mempunyai jala kromatin lebih padat
dari basofilik eritroblas, dan selnya lebih kecil.- d. Ortokromatik Eritroblas (Normoblas)- Polikromatik Eritroblas membelah beberapa kali secara mitosis. Normoblas lebih kecil
daripada Polikromatik Eritroblas dan mengandung inti yang lebih kecil yang terwarnai
basofil padat. Intinya secara bertahap menjadi piknotik. Tidak ada lagi aktivitas mitosis.
Akhirnya inti dikeluarkan dari sel bersama-sama dengan pinggiran tipis sitoplasma. Inti
yang sudah dikeluarkan dimakan oleh makrofagmakrofag- yang ada di dalam stroma sumsum tulang- e. Retikulosit

29
- Retikulosit adalah sel-sel eritrosit muda yang kehilangan inti selnya, dan mengandung
sisa-sisa asam ribonukleat di dalam sitoplasmanya, serta masih dapat mensintesis
hemoglobin. Retikulosit dianggap kehilangan sumsum retikularnya sebelum
meninggalkan sumsum tulang, karena jumlah retikulosit dalam darah perifer normal
kurang dari satu persen dari jumlah eritrosit. Dalam keadaan normal keempat tahap
pertama sebelum menjadi retikulosit terdapat pada sumsung tulang. Retikulosit terdapat
baik pada sumsum tulang maupun darah tepi. Di dalam sumsum tulang memerlukan
waktu kurang lebih 2 – 3 hari untuk menjadi matang, sesudah itu lepas ke dalam darah. - f. Eritrosit- Eritrosit merupakan produk akhir dari perkembangan eritropoesis. Sel ini berbentuk
lempengan bikonkaf dan dibentuk di sumsum tulang. Pada manusia, sel ini berada di
dalam sirkulasi selama kurang lebih 120- hari. Jumlah normal pada tubuh laki – laki 5,4 juta/µl dan pada perempuan 4,8 juta/µl.
setiap eritrosit memiliki diameter sekitar 7,5 µm dan tebal 2 µm. - Perkembangan normal eritrosit tergantung pada banyak macammacam faktor, termasuk
adanya substansi asal (terutama globin, hem dan besi). Faktor-faktor lain, seperti asam
askorbat, vitamin B12, dan faktor intrinsic (normal ada dalam getah lamung), yang
berfungsi sebagai koenzim pada proses sintesis, juga penting untuk pendewasaan normal
eritrosit.- Pada sistem Eritropoesis dikenal juga istilah Eritropoiesis inefektif, yang dimaksud
Eritropoiesis inefektif adalah suatu proses penghancuran sel induk eritroid yang prematur
disumsum tulang. Choi, dkk, dalam- studinya bahwa pengukuran radio antara retikulosit di sumsum tulang terhadap retikulosit
di darah tepi merupakan ukuran yang pentng untuk bisa memperkirakan beratnya
gangguan produksi SDM. - 2. Seri Leukosit- a. Leukosit Granulosit / myelosit- Myelosit terdiri dari 3 jenis yaitu neutrofil, eosinofil dan basofil yang mengandung
granula spesifik yang khas. Tahapan perkembangan myelosit yaitu :- 1) Mieloblas

30
- Mieloblas adalah sel yang paling muda yang dapat dikenali dari seri granulosit. Diameter
berkisar antara 10-15µm. Intinya yang bulat dan besar memperlihatkan kromatin halus
serta satu atau dua anak inti.- 2) Promielosit- Sel ini agak lebih besar dari mielobas. Intinya bulat atau lonjong, serta anak inti yang tak
jelas.- 3) Mielosit- Promielosit berpoliferasi dan berdiferensiasi menjadi mielosit. Pada proses diferensiasi
timbul grnula spesifik, dengan ukuran, bentuk, dan sifat terhadap pewarnaan yang
memungkinkan seseorang mengenalnya sebagai neutrofil, eosinofil, atau basofil.
Diameter berkisar 10µm, inti mengadakan cekungan dan mulai berbentuk seperti tapal
kuda.- 4) Metamielosit- Setelah mielosit membelah berulang-ulang, sel menjadi lebih kecil kemudian berhenti
membelah. Sel-sel akhir pembelahan adalah metamielosit. Metamielosit mengandung
granula khas, intinya berbentuk cekungan. Pada akhir tahap ini, metamielosit dikenal
sebagai sel batang. Karena sel-sel bertambah tua, inti berubah, membentuk lobus khusus
dan jumlah lobi bervariasi dari 3 sampai 5. Sel dewasa (granulosit bersegmen) masuk
sinusoid-sinusoid dan mencapai peredaran darah. Pada masing-masing tahap mielosit
yang tersebut di atas jumlah neutrofil jauh lebih banyak daripada eosinofil dan basofil.- b. Leukosit non granuler- 1) Limfosit- Sel-sel precursor limfosit adalah limfoblas, yang merupakan sel berukuran relatif besar,
berbentuk bulat. Intinya besar dan mengandung kromatin yang relatif dengan anak inti
mencolok. Sitoplasmanya homogen dan basofil. Ketika limfoblas mengalami diferensiasi,
kromatin intinya menjadi lebih tebal dan padat dan granula azurofil terlihat dalam
sitoplasma. Ukuran selnya berkurang dan diberi nama prolimfosit. Sel-sel tersebut
langsung menjadi limfosit yang beredar.- 2) Monosit
31
- Monosit awalnya adalah monoblas berkembang menjadi promonosit. Sel ini berkembang
menjadi monosit. Monosit meninggalkan darah lalu masuk ke jaringan, disitu jangka
hidupnya sebagai makrofag mungkin 70 hari.- 3. Seri Trombosit (Trombopoesis)- Pembentukan Megakariosit dan Keping-keping darah Megakariosit adalah sel raksasa
(diameter 30-100µm atau lebih). Inti berlobi secara kompleks dan dihubungkan dengan
benang-benang halus dari bahan kromatin. Sitoplasma mengandung banyak granula
azurofil dan memperlihatkan sifat basofil setempat. Megakariosit membentuk
tonjolantonjolan sitoplasma yang akan dilepas sebagai keping-keping darah. Setelah
sitoplasma perifer lepas sebagai keping-keping darah, megakariosit mengeriput dan
intinya hancur.
2. Thalasemia
Thalassemia adalah sekelompok anemia hipokromik herediter dengan berbagai derajat
keparahan. Defek genetik yang mendasari meliputi delesi total atau parsial gen globin dan
substitusi, delesi, atau insersi nukleotida. Akibat dari berbagai perubahan ini adalah penurunan
atau tidak adanya mRNA bagi satu atau lebih rantai globin atau pembentukan mRNA yang cacat
secara fungsional. Akibatnya adalah penurunan dan supresi total sintesis rantai polipeptida Hb.
Kira-kira 100 mutasi yang berbeda telah ditemukan mengakibatkan fenotip thalassemia; banyak
di antara mutasi ini adalah unik untuk daerah geografi setempat. Pada umumnya, rantai globin
yang disintesis dalam eritrosit thalassemia secara struktural adalah normal. Pada bentuk
thalassemia-α yang berat, terbentuk hemoglobin hemotetramer abnormal (β4 atau γ4) tetapi
komponen polipeptida globin mempunyai struktur normal. Sebaliknya, sejumlah Hb abnormal
juga menyebabkan perubahan hemotologi mirip thalassemia.
Gen thalassemia sangat luas tersebar, dan kelainan ini diyakini merupakan penyakit
genetik manusia yang paling prevalen. Distribusi utama meliputi daerah-daerah perbatasan Laut

32
Mediterania, sebagian besar Afrika, Timur Tengah, sub-benua India, dan Asia Tenggara. Dari
3% sampai 8% orang Amerika keturunan Itali atau Yunani dan 0,5 % dari kulit hitam Amerika
membawa gen untuk thalassemia-β. Di beberapa daerah Asia Tenggara sebanyak 40 % dari
populasi mempunyai satu atau lebih gen thalassemia.
A. Epidemiologi
Di seluruh dunia, 15 juta orang memiliki presentasi klinis dari thalassemia. Fakta ini
mendukung thalassemia sebagai salah satu penyakit turunan yang terbanyak; menyerang hampir
semua golongan etnik dan terdapat pada hampir seluruh negara di dunia.
Beberapa tipe thalassemia lebih umum terdapat pada area tertentu di dunia. Thalassemia-
β lebih sering ditemukan di negara-negara Mediteraniam seperti Yunani, Itali, dan Spanyol.
Banyak pulau-pulau Mediterania seperti Ciprus, Sardinia, dan Malta, memiliki insidens
thalassemia-β mayor yang tinggi secara signifikan. Thalassemia-β juga umum ditemukan di
Afrika Utara, India, Timur Tengah, dan Eropa Timur. Sebaliknya, thalassemia-α lebih sering
ditemukan di Asia Tenggara, India, Timur Tengah, dan Afrika.
Mortalitas dan Morbiditas
Thalassemia-α mayor adalah penyakit yang mematikan, dan semua janin yang terkena
akan lahir dalam keadaan hydrops fetalis akibat anemia berat. Beberapa laporan pernah
mendeskripsikan adanya neonatus dengan thalassemia-α mayor yang bertahan setelah mendapat
transfusi intrauterin. Penderita seperti ini membutuhkan perawatan medis yang ekstensif
setelahnya, termasuk transfusi darah teratur dan terapi khelasi, sama dengan penderita
thalassemia-β mayor. Terdapat juga laporan kasus yang lebih jarang mengenai neonatus dengan
thalassemia-α mayor yang lahir tanpa hydrops fetalis yang bertahan tanpa transfusi intrauterin.

33
Pada kasus ini, tingginya level Hb Portland, yang merupakan Hb fungsional embrionik,
diperkirakan sebagai penyebab kondisi klinis yang jarang tersebut.
Pada pasien dengan berbagai tipe thalassemia-β, mortalitas dan morbiditas bervariasi
sesuai tingkat keparahan dan kualitas perawatan. Thalassemia-β mayor yang berat akan berakibat
fatal bila tidak diterapi. Gagal jantung akibat anemia berat atau iron overload adalah penyebab
tersering kematian pada penderita. Penyakit hati, infeksi fulminan, atau komplikasi lainnya yang
dicetuskan oleh penyakit ini atau terapinya termasuk merupakan penyebab mortalitas dan
morbiditas pada bentuk thalassemia yang berat.
Mortalitas dan morbiditas tidak terbatas hanya pada penderita yang tidak diterapi; mereka
yang mendapat terapi yang dirancang dengan baik tetap berisiko mengalami bermacam-macam
komplikasi. Kerusakan organ akibat iron overload, infeksi berat yang kronis yang dicetuskan
transfusi darah, atau komplikasi dari terapi khelasi, seperti katarak, tuli, atau infeksi, merupakan
komplikasi yang potensial.
Usia
Meskipun thalassemia merupakan penyakit turunan (genetik), usia saat timbulnya gejala
bervariasi secara signifikan. Dalam talasemia, kelainan klinis pada pasien dengan kasus-kasus
yang parah dan temuan hematologikpada pembawa (carrier) tampak jelas pada saat lahir.
Ditemukannya hipokromia dan mikrositosis yang tidak jelas penyebabnya pada neonatus,
digambarkan di bawah ini, sangat mendukung diagnosis.

34
Gambar 1. Sapuan apus darah tepi Penyakit Hb H pada neonatus
Namun, pada thalassemia-β berat, gejala mungkin tidak jelas sampai paruh kedua tahun
pertama kehidupan; sampai waktu itu, produksi rantai globin γ dan penggabungannya ke Hb
Fetal dapat menutupi gejala untuk sementara.
Bentuk thalassemia ringan sering ditemukan secara kebetulan pada berbagai usia. Banyak
pasien dengan kondisi thalassemia-β homozigot yang jelas (yaitu, hipokromasia, mikrositosis,
elektroforesis negatif untuk Hb A, bukti bahwa kedua orang tua terpengaruh) mungkin tidak
menunjukkan gejala atau anemia yang signifikan selama beberapa tahun. Hampir semua pasien
dengan kondisi tersebut dikategorikan sebagai thalassemia-β intermedia. Situasi ini biasanya
terjadi jika pasien mengalami mutasi yang lebih ringan.
B. Patofisiologi
Thalassemia adalah kelainan herediter dari sintesis Hb akibat dari gangguan produksi
rantai globin. Penurunan produksi dari satu atau lebih rantai globin tertentu (α,β,γ,δ) akan
menghentikan sintesis Hb dan menghasilkan ketidakseimbangan dengan terjadinya produksi
rantai globin lain yang normal.
Karena dua tipe rantai globin (α dan non-α) berpasangan antara satu sama lain dengan
rasio hampir 1:1 untuk membentuk Hb normal, maka akan terjadi produksi berlebihan dari
rantai globin yang normal dan terjadi akumulasi rantai tersebut di dalam sel menyebabkan sel

35
menjadi tidak stabil dan memudahkan terjadinya destruksi sel. Ketidakseimbangan ini
merupakan suatu tanda khas pada semua bentuk thalassemia. Karena alasan ini, pada sebagian
besar thalassemia kurang sesuai disebut sebagai hemoglobinopati karena pada tipe-tipe
thalassemia tersebut didapatkan rantai globin normal secara struktural dan juga karena defeknya
terbatas pada menurunnya produksi dari rantai globin tertentu.
Tipe thalassemia biasanya membawa nama dari rantai yang tereduksi. Reduksi bervariasi
dari mulai sedikit penurunan hingga tidak diproduksi sama sekali (complete absence). Sebagai
contoh, apabila rantai β hanya sedikit diproduksi, tipe thalassemia-nya dinamakan sebagai
thalassemia-β+, sedangkan tipe thalassemia-β° menandakan bahwa pada tipe tersebut rantai β
tidak diproduksi sama sekali. Konsekuensi dari gangguan produksi rantai globin mengakibatkan
berkurangnya deposisi Hb pada sel darah merah (hipokromatik). Defisiensi Hb menyebabkan sel
darah merah menjadi lebih kecil, yang mengarah ke gambaran klasik thalassemia yaitu anemia
hipokromik mikrositik. Hal ini berlaku hampir pada semua bentuk anemia yang disebabkan oleh
adanya gangguan produksi dari salah satu atau kedua komponen Hb : heme atau globin. Namun
hal ini tidak terjadi pada silent carrier, karena pada penderita ini jumlah Hb dan indeks sel darah
merah berada dalam batas normal.
Pada tipe trait thalassemia-β yang paling umum, level Hb A2 (δ2/α2) biasanya meningkat.
Hal ini disebabkan oleh meningkatnya penggunaan rantai δ oleh rantai α bebas yang eksesif,
yang mengakibatkan terjadinya kekurangan rantai β adekuat untuk dijadikan pasangan. Gen δ,
tidak seperti gen β dan α, diketahui memiliki keterbatasan fisiologis dalam kemampuannya untuk
memproduksi rantai δ yang stabil; dengan berpasangan dengan rantai α, rantai δ memproduksi
Hb A2 (kira-kira 2,5-3% dari total Hb). Sebagian dari rantai α yang berlebihan digunakan untuk
membentuk Hb A2, dimana sisanya (rantai α) akan terpresipitasi di dalam sel, bereaksi dengan

36
membran sel, mengintervensi divisi sel normal, dan bertindak sebagai benda asing sehingga
terjadinya destruksi dari sel darah merah. Tingkat toksisitas yang disebabkan oleh rantai yang
berlebihan bervariasi berdasarkan tipe dari rantai itu sendiri (misalnya toksisitas dari rantai α
pada thalassemia-β lebih nyata dibandingkan toksisitas rantai β pada thalassemia-α).
Dalam bentuk yang berat, seperti thalassemia-β mayor atau anemia Cooley, berlaku
patofisiologi yang sama dimana terdapat adanya substansial yang berlebihan. Kelebihan rantai α
bebas yang signifikan akibat kurangnya rantai β akan menyebabkan terjadinya pemecahan
prekursor sel darah merah di sumsum tulang (eritropoesis inefektif).
Produksi Rantai Globin
Untuk memahami perubahan genetik pada thalassemia, kita perlu mengenali dengan baik
proses fisiologis dari produksi rantai globin pada orang sehat atau normal. Suatu unit rantai
globin merupakan komponen utama untuk membentuk Hb : bersama-sama dengan Heme, rantai
globin menghasilkan Hb. Dua pasangan berbeda dari rantai globin akan membentuk struktur
tetramer dengan Heme sebagai intinya. Semua Hb normal dibentuk dari dua rantai globin α (atau
mirip-α) dan dua rantai globin non-α. Bermacam-macam tipe Hb terbentuk, tergantung dari tipe
rantai globin yang membentuknya. Masing-masing tipe Hb memiliki karakteristik yang berbeda
dalam mengikat oksigen, biasanya berhubungan dengan kebutuhan oksigen pada tahap-tahap
perkembangan yang berbeda dalam kehidupan manusia.
Pada masa kehidupan embrionik, rantai ζ(rantai mirip-α) berkombinasi dengan rantai γ
membentuk Hb Portland (ζ2γ2) dan dengan rantai ε untuk membentuk Hb Gower-1 (ζ2ε2).

37
Selanjutnya, ketika rantai α telah diproduksi, dibentuklah Hb Gower-2, berpasangan dengan
rantai ε (α2ε2). Hb Fetal dibentuk dari α2γ2 dan Hb dewasa primer (Hb A) dibentuk dari α2β2. Hb
fisiologis yang ketiga, Hb A2, dibentuk dari rantai α2δ2.
Gambar 2. Gen rantai α yang berduplikasi pada kromosom 16 berpasangan dengan rantai-rantai non-α untuk memproduksi bermacam-macam Hb normal.
Patofisiologi seluler
Kelainan dasar dari semua tipe thalassemia adalah ketidakseimbangan sintesis rantai
globin. Namun, konsekuensi akumulasi dari produksi rantai globin yang berlebihan berbeda-beda
pada tiap tipe thalassemia. Pada thalassemia-β, rantai α yang berlebihan, tidak mampu
membentuk Hb tetramer, terpresipitasi di dalam prekursor sel darah merah dan, dengan berbagai
cara, menimbulkan hampir semua gejala yang bermanifestasi pada sindroma thalassemia-β;
situasi ini tidak terjadi pada thalassemia-α.
Rantai globin yang berlebihan pada thalassemia-α adalah rantai γ pada tahun-tahun
pertama kehidupan, dan rantai β pada usia yang lebih dewasa. Rantai-rantai tipe ini relatif
bersifat larut sehingga mampu membentuk homotetramer yang, meskipun relatif tidak stabil,
mampu tetap bertahan (viable) dan dapat memproduksi molekul Hb seperti Hb Bart (γ4) dan Hb

38
H (β4). Perbedaan dasar pada dua tipe utama ini mempengaruhi perbedaan besar pada manifestasi
klinis dan tingkat keparahan dari penyakit ini.
Rantai α yang terakumulasi di dalam prekursor sel darah merah bersifat tidak larut
(insoluble), terpresipitasi di dalam sel, berinteraksi dengan membran sel (mengakibatkan
kerusakan yang signifikan), dan mengganggu divisi sel. Kondisi ini menyebabkan terjadinya
destruksi intramedular dari prekursor sel darah merah. Sebagai tambahan, sel-sel yang bertahan
yang sampai ke sirkulasi darah perifer dengan intracellular inclusion bodies (rantai yang
berlebih) akan mengalami hemolisis; hal ini berarti bahwa baik hemolisis maupun eritropoesis
inefektif menyebabkan anemia pada penderita dengan thalassemia-β.
Kemampuan sebagian sel darah merah untuk mempertahankan produksi dari rantai γ,
yang mampu untuk berpasangan dengan sebagian rantai α yang berlebihan untuk membentuk Hb
F, adalah suatu hal yang menguntungkan. Ikatan dengan sebagian rantai berlebih tidak diragukan
lagi dapat mengurangi gejala dari penyakit dan menghasilkan Hb tambahan yang memiliki
kemampuan untuk membawa oksigen.
Selanjutnya, peningkatan produksi Hb F sebagai respon terhadap anemia berat,
menimbulkan mekanisme lain untuk melindungi sel darah merah pada penderita dengan
thalassemia-β. Peningkatan level Hb F akan meningkatkan afinitas oksigen, menyebabkan
terjadinya hipoksia, dimana, bersama-sama dengan anemia berat akan menstimulasi produksi
dari eritropoetin. Akibatnya, ekspansi luas dari massa eritroid yang inefektif akan menyebabkan
ekspansi tulang berat dan deformitas. Baik penyerapan besi dan laju metabolisme akan
meningkat, berkontribusi untuk menambah gejala klinis dan manifestasi laboratorium dari
penyakit ini. Sel darah merah abnormal dalam jumlah besar akan diproses di limpa, yang

39
bersama-sama dengan adanya hematopoesis sebagai respon dari anemia yang tidak diterapi, akan
menyebabkan splenomegali masif yang akhirnya akan menimbulkan terjadinya hipersplenisme.
Apabila anemia kronik pada penderita dikoreksi dengan transfusi darah secara teratur,
maka ekspansi luas dari sumsum tulang akibat eritropoesis inefektif dapat dicegah atau
dikembalikan seperti semula. Memberikan sumber besi tambahan secara teori hanya akan lebih
merugikan pasien. Namun, hal ini bukanlah masalah yang sebenarnya, karena penyerapan besi
diregulasi oleh dua faktor utama : eritropoesis inefektif dan jumlah besi pada penderita yang
bersangkutan. Eritropoesis yang inefektif akan menyebabkan peningkatan absorpsi besi karena
adanya downregulation dari gen HAMP, yang memproduksi hormon hepar yang dinamakan
hepcidin, regulator utama pada absorpsi besi di usus dan resirkulasi besi oleh makrofag. Hal ini
terjadi pada penderita dengan thalassemia intermedia.
Dengan pemberian transfusi darah, eritropoesis yang inefektif dapat diperbaiki, dan
terjadi peningkatan jumlah hormon hepcidin; sehingga penyerapan besi akan berkurang dan
makrofag akan mempertahankan kadar besi.
Pada pasien dengan iron overload (misalnya hemokromatosis), absorpsi besi menurun
akibat meningkatnya jumlah hepsidin. Namun, hal ini tidak terjadi pada penderita thalassemia-β
berat karena diduga faktor plasma menggantikan mekanisme tersebut dan mencegah terjadinya
produksi hepsidin sehingga absorpsi besi terus berlangsung meskipun penderita dalam keadaan
iron overload.
Efek hepsidin terhadap siklus besi dilakukan melalui kerja hormon lain bernama
ferroportin, yang mentransportasikan besi dari enterosit dan makrofag menuju plasma dan
menghantarkan besi dari plasenta menuju fetus. Ferroportin diregulasi oleh jumlah penyimpanan
besi dan jumlah hepsidin. Hubungan ini juga menjelaskan mengapa penderita dengan

40
thalassemia-β yang memiliki jumlah besi yang sama memiliki jumlah ferritin yang berbeda
sesuai dengan apakah mereka mendapat transfusi darah teratur atau tidak. Sebagai contoh,
penderita thalassemia-β intermedia yang tidak mendapatkan transfusi darah memiliki jumlah
ferritin yang lebih rendah dibandngkan dengan penderita yang mendapatkan transfusi darah
secara teratur, meskipun keduanya memiliki jumlah besi yang sama.
Kebanyakan besi non-heme pada individu yang sehat berikatan kuat dengan protein
pembawanya, transferrin. Pada keadaan iron overload, seperti pada thalassemia berat, transferrin
tersaturasi, dan besi bebas ditemukan di plasma. Besi ini cukup berbahaya karena memiliki
material untuk memproduksi hidroksil radikal dan akhirnya akan terakumulasi pada organ-organ,
seperti jantung, kelenjar endokrin, dan hati, mengakibatkan terjadinya kerusakan pada organ-
organ tersebut (organ damage).
Hipotesa Malaria
Pada tahun 1949, Haldane menyatakan adanya suatu keuntungan selektif untuk bertahan
hidup pada individu dengan trait thalassemia pada daerah endemik malaria. Hardane berpendapat
bahwa penyakit sel darah merah letal seperti pada thalassemia, anemia sel sabit, dan defisiensi
G6PD terdapat hampir secara eksklusif pada daerah tropis dan subtropis. Insidens dari mutasi
genetik ini pada populas tertentu merefleksikan adanya keseimbangan antara kematian dini pada
penderita homozigot dengan peningkatan kesehatan pada penderita heterozigot.
Mekanisme proteksi terhadap malaria pada penderita trait thalassemia belum jelas. Sel
Hb F telah didemonstrasikan dapat menghambat pertumbuhan parasit malaria, dan, berdasarkan
tingginya level Hb F tersebut pada bayi dengan trait thalassemia-β, malaria serebral fatal yang
diketahui dapat menyebabkan kematian pada bayi tersebut dapat dicegah. Sel darah merah pada

41
penderita Penyakit Hb H juga memiliki semacam efek supresif terhadap pertumbuhan parasit.
Namun efek ini tidak ditemukan pada penderita dengan trait thalassemia-α.
C. Klasifikasi Thalassemia dan Presentasi Klinisnya
Saat ini dikenal sejumlah besar sindrom thalasemia; masing-masing melibatkan penurunan
produksi satu atau lebih rantai globin, yang membentuk bermacam-macam jenis Hb yang
ditemukan pada sel darah merah. Jenis yang paling penting dalam praktek klinis adalah sindrom
yang mempengaruhi baik atau sintesis rantai α maupun β.
Thalassemia-α
Anemia mikrositik yang disebabkan oleh defisiensi sintesis globin-α banyak ditemukan
di Afrika, negara di daerah Mediterania, dan sebagian besar Asia. Delesi gen globin-α
menyebabkan sebagian besar kelainan ini. Terdapat empat gen globin-α pada individu normal,
dan empat bentuk thalassemia-α yang berbeda telah diketahui sesuai dengan delesi satu, dua,
tiga, dan semua empat gen ini
Tabel 1. Thalassemia-α
G e n o t i p J u m l a h g e n α Presentasi Klinis H e m o g l o b i n E l e k t r o f o r e s i s
S a a t L a h i r > 6 b u l a n
α α / α α 4 N o r m a l N N
- α / α α 3 S i l e n t c a r r i e r 0-3 % Hb Bar t s N
--/αα atau –α/-α 2 T r a i t t h a l - α 2-10% Hb Barts N
- - / - α 1 P e n y a k i t H b H 15-30% Hb Bart H b H

42
- - / - - 0 Hydrops fe tal i s > 7 5 % H b B a r t -
Ket : N = hasil normal, Hb = hemoglobin, Hb Bart’s = γ4, HbH = β4
Silent carrier thalassemia-α
o Merupakan tipe thalassemia subklinik yang paling umum, biasanya ditemukan
secara kebetulan diantara populasi, seringnya pada etnik Afro-Amerika. Seperti
telah dijelaskan sebelumnya, terdapat 2 gen α yang terletak pada kromosom 16.
o Pada tipe silent carrier, salah satu gen α pada kromosom 16 menghilang,
menyisakan hanya 3 dari 4 gen tersebut. Penderita sehat secara hematologis,
hanya ditemukan adanya jumlah eritrosit (sel darah merah) yang rendah dalam
beberapa pemeriksaan.
o Pada tipe ini, diagnosis tidak dapat dipastikan dengan pemeriksaan elektroforesis
Hb, sehingga harus dilakukan tes lain yang lebih canggih. Bisa juga dicari akan
adanya kelainan hematologi pada anggota keluarga ( misalnya orangtua) untuk
mendukung diagnosis. Pemeriksaan darah lengkap pada salah satu orangtua yang
menunjukkan adanya hipokromia dan mikrositosis tanpa penyebab yang jelas
merupakan bukti yang cukup kuat menuju diagnosis thalasemia.
Trait thalassemia-α
o Trait ini dikarakterisasi dengan anemia ringan dan jumlah sel darah merah yang
rendah. Kondisi ini disebabkan oleh hilangnya 2 gen α pada satu kromosom 16
atau satu gen α pada masing-masing kromosom. Kelainan ini sering ditemukan di
Asia Tenggara, subbenua India, dan Timur Tengah.

43
o Pada bayi baru lahir yang terkena, sejumlah kecil Hb Barts (γ4) dapat ditemukan
pada elektroforesis Hb. Lewat umur satu bulan, Hb Barts tidak terlihat lagi, dan
kadar Hb A2 dan HbF secara khas normal.
Gambar 3. Thalassemia alpha menurut hukum Mendel
Penyakit Hb H
o Kelainan disebabkan oleh hilangnya 3 gen globin α, merepresentasikan
thalassemia-α intermedia, dengan anemia sedang sampai berat, splenomegali,
ikterus, dan jumlah sel darah merah yang abnormal. Pada sediaan apus darah tepi
yang diwarnai dengan pewarnaan supravital akan tampak sel-sel darah merah
yang diinklusi oleh rantai tetramer β (Hb H) yang tidak stabil dan terpresipitasi di
dalam eritrosit, sehingga menampilkan gambaran golf ball. Badan inklusi ini
dinamakan sebagai Heinz bodies.

44
Gambar 4. Pewarnaan supravital pada sapuan apus darah tepi Penyakit Hb H yang menunjukkan Heinz-Bodies
Thalassemia-α mayor
o Bentuk thalassemia yang paling berat, disebabkan oleh delesi semua gen globin-α,
disertai dengan tidak ada sintesis rantai α sama sekali.
o Karena Hb F, Hb A, dan Hb A2 semuanya mengandung rantai α, maka tidak
satupun dari Hb ini terbentuk. Hb Barts (γ4) mendominasi pada bayi yang
menderita, dan karena γ4 memiliki afinitas oksigen yang tinggi, maka bayi-bayi
itu mengalami hipoksia berat. Eritrositnya juga mengandung sejumlah kecil Hb
embrional normal (Hb Portland = ζ2γ2), yang berfungsi sebagai pengangkut
oksigen.
o Kebanyakan dari bayi-bayi ini lahir mati, dan kebanyakan dari bayi yang lahir
hidup meninggal dalam waktu beberapa jam. Bayi ini sangat hidropik, dengan
gagal jantung kongestif dan edema anasarka berat. Yang dapat hidup dengan
manajemen neonatus agresif juga nantinya akan sangat bergantung dengan
transfusi.
Thalassemia-β

45
Sama dengan thalassemia-α, dikenal beberapa bentuk klinis dari thalassemia-β; antara
lain :
Silent carrier thalassemia-β
o Penderita tipe ini biasanya asimtomatik, hanya ditemukan nilai eritrosit yang
rendah. Mutasi yang terjadi sangat ringan, dan merepresentasikan suatu
thalassemia-β+.
o Bentuk silent carrier thalassemia-β tidak menimbulkan kelainan yang dapat
diidentifikasi pada individu heterozigot, tetapi gen untuk keadaan ini, jika
diwariskan bersama-sama dengan gen untuk thalassemia-β°, menghasilkan
sindrom thalassemia intermedia.
Gambar 5. Thalassemia beta menurut Hukum Mendel
Trait thalassemia-β
o Penderita mengalami anemia ringan, nilai eritrosit abnormal, dan elektroforesis
Hb abnormal dimana didapatkan peningkatan jumlah Hb A2, Hb F, atau keduanya

46
o Individu dengan ciri (trait) thalassemia sering didiagnosis salah sebagai anemia
defisiensi besi dan mungkin diberi terapi yang tidak tepat dengan preparat besi
selama waktu yang panjang. Lebih dari 90% individu dengan trait thalassemia-β
mempunyai peningkatan Hb-A2 yang berarti (3,4%-7%). Kira-kira 50% individu
ini juga mempunyai sedikit kenaikan HbF, sekitar 2-6%. Pada sekelompok kecil
kasus, yang benar-benar khas, dijumpai Hb A2 normal dengan kadar HbF berkisar
dari 5% sampai 15%, yang mewakili thalassemia tipe δβ.
Thalassemia-β yang terkait dengan variasi struktural rantai β
o Presentasi klinisnya bervariasi dari seringan thalassemia media hingga seberat
thalassemia-β mayor
o Ekspresi gen homozigot thalassemia (β+) menghasilkan sindrom mirip anemia
Cooley yang tidak terlalu berat (thalassemia intermedia). Deformitas skelet dan
hepatosplenomegali timbul pada penderita ini, tetapi kadar Hb mereka biasanya
bertahan pada 6-8 gr/dL tanpa transfusi.
o Kebanyakan bentuk thalassemia-β heterozigot terkait dengan anemia ringan.
Kadar Hb khas sekitar 2-3 gr/dL lebih rendah dari nilai normal menurut umur.

47
o Eritrosit adalah mikrositik hipokromik dengan poikilositosis, ovalositosis, dan
seringkali bintik-bintik basofil. Sel target mungkin juga ditemukan tapi biasanya
tidak mencolok dan tidak spesifik untuk thalassemia.
o MCV rendah, kira-kira 65 fL, dan MCH juga rendah (<26 pg). Penurunan ringan
pada ketahanan hidup eritrosit juga dapat diperlihatkan, tetapi tanda hemolisis
biasanya tidak ada. Kadar besi serum normal atau meningkat.
Thalassemia-β° homozigot (Anemia Cooley, Thalassemia Mayor)
o bergejala sebagai anemia hemolitik kronis yang progresif selama 6 bulan kedua
kehidupan. Transfusi darah yang reguler diperlukan pada penderita ini untuk
mencegah kelemahan yang amat sangat dan gagal jantung yang disebabkan oleh
anemia. Tanpa transfusi, 80% penderita meninggal pada 5 tahun pertama
kehidupan.
o Pada kasus yang tidak diterapi atau pada penderita yang jarang menerima
transfusi pada waktu anemia berat, terjadi hipertrofi jaringan eritropoetik
disumsum tulang maupun di luar sumsum tulang. Tulang-tulang menjadi tipis dan
fraktur patologis mungkin terjadi. Ekspansi masif sumsum tulang di wajah dan
tengkorak menghasilkan bentuk wajah yang khas.

48
Gambar 6. Deformitas tulang pada thalassemia beta mayor (Facies Cooley)
o Pucat, hemosiderosis, dan ikterus sama-sama memberi kesan coklat kekuningan.
Limpa dan hati membesar karena hematopoesis ekstrameduler dan hemosiderosis.
Pada penderita yang lebih tua, limpa mungkin sedemikian besarnya sehingga
menimbulkan ketidaknyamanan mekanis dan hipersplenisme sekunder.
Gambar 7. Splenomegali pada thalassemia
o Pertumbuhan terganggu pada anak yang lebih tua; pubertas terlambat atau tidak
terjadi karena kelainan endokrin sekunder. Diabetes mellitus yang disebabkan

49
oleh siderosis pankreas mungkin terjadi. Komplikasi jantung, termasuk aritmia
dan gagal jantung kongestif kronis yang disebabkan oleh siderosis miokardium
sering merupakan kejadian terminal.
o Kelainan morfologi eritrosit pada penderita thalassemia-β° homozigot yang tidak
ditransfusi adalah ekstrem. Disamping hipokromia dan mikrositosis berat, banyak
ditemukan poikilosit yang terfragmentasi, aneh (sel bizarre) dan sel target.
Sejumlah besar eritrosit yang berinti ada di darah tepi, terutama setelah
splenektomi. Inklusi intraeritrositik, yang merupakan presipitasi kelebihan rantai
α, juga terlihat pasca splenektomi. Kadar Hb turun secara cepat menjadi < 5 gr/dL
kecuali mendapat transfusi. Kadar serum besi tinggi dengan saturasi kapasitas
pengikat besi (iron binding capacity). Gambaran biokimiawi yang nyata adalah
adanya kadar HbF yang sangat tinggi dalam eritrosit.
D. Stadium Thalassemia
Terdapat suatu sistem pembagian stadium thalassemia berdasarkan jumlah kumulatif
transfusi darah yang diberikan pada penderita untuk menentukan tingkat gejala yang melibatkan
kardiovaskuler dan untuk memutuskan kapan untuk memulai terapi khelasi pada pasien dengan
thalassemia-β mayor atau intermedia. Pada sistem ini, pasien dibagi menjadi tiga kelompok,
yaitu :
Stadium I
o Merupakan mereka yang mendapat transfusi kurang dari 100 unit Packed Red
Cells (PRC). Penderita biasanya asimtomatik, pada echokardiogram (ECG) hanya

50
ditemukan sedikit penebalan pada dinding ventrikel kiri, dan elektrokardiogram
(EKG) dalam 24 jam normal.
Stadium II
o Merupakan mereka yang mendapat transfusi antara 100-400 unit PRC dan
memiliki keluhan lemah-lesu. Pada ECG ditemukan penebalan dan dilatasi pada
dinding ventrikel kiri. Dapat ditemukan pulsasi atrial dan ventrikular abnormal
pada EKG dalam 24 jam
Stadium III
o Gejala berkisar dari palpitasi hingga gagal jantung kongestif, menurunnya fraksi
ejeksi pada ECG. Pada EKG dalam 24 jam ditemukan pulsasi prematur dari atrial
dan ventrikular.
E. Terapi
Penderita trait thalassemia tidak memerlukan terapi ataupun perawatan lanjut setelah
diagnosis awal dibuat. Terapi preparat besi sebaiknya tidak diberikan kecuali memang dipastikan
terdapat defisiensi besi dan harus segera dihentikan apabila nilai Hb yang potensial pada
penderita tersebut telah tercapai. Diperlukan konseling pada semua penderita dengan kelainan
genetik, khususnya mereka yang memiliki anggota keluarga yang berisiko untuk terkena
penyakit thalassemia berat.
Penderita thalassemia berat membutuhkan terapi medis, dan regimen transfusi darah
merupakan terapi awal untuk memperpanjang masa hidup. Transfusi darah harus dimulai pada
usia dini ketika anak mulai mengalami gejala dan setelah periode pengamatan awal untuk
menilai apakah anak dapat mempertahankan nilai Hb dalam batas normal tanpa transfusi.
Transfusi Darah

51
Transfusi darah bertujuan untuk mempertahankan nilai Hb tetap pada level 9-9.5 gr/dL
sepanjang waktu.
Pada pasien yang membutuhkan transfusi darah reguler, maka dibutuhkan suatu studi
lengkap untuk keperluan pretransfusi. Pemeriksaan tersebut meliputi fenotip sel darah
merah, vaksinasi hepatitis B (bila perlu), dan pemeriksaan hepatitis.
Darah yang akan ditransfusikan harus rendah leukosit; 10-15 mL/kg PRC dengan
kecepatan 5 mL/kg/jam setiap 3-5 minggu biasanya merupakan regimen yang adekuat
untuk mempertahankan nilai Hb yang diinginkan.
Pertimbangkan pemberikan asetaminofen dan difenhidramin sebelum transfusi untuk
mencegah demam dan reaksi alergi.
Komplikasi Transfusi Darah
Komplikasi utama dari transfusi adalah yang berkaitan dengan transmisi bahan infeksius
ataupun terjadinya iron overload. Penderita thalassemia mayor biasanya lebih mudah untuk
terkena infeksi dibanding anak normal, bahkan tanpa diberikan transfusi. Beberapa tahun lalu,
25% pasien yang menerima transfusi terekspose virus hepatitis B. Saat ini, dengan adanya
imunisasi, insidens tersebut sudah jauh berkurang. Virus Hepatitis C (HCV) merupakan
penyebab utama hepatitis pada remaja usia di atas 15 tahun dengan thalassemia. Infeksi oleh
organisme opurtunistik dapat menyebabkan demam dan enteriris pada penderita dengan iron
overload, khususnya mereka yang mendapat terapi khelasi dengan Deferoksamin (DFO).
Demam yang tidak jelas penyebabnya, sebaiknya diterapi dengan Gentamisin dan Trimetoprim-
Sulfametoksazol.

52
Terapi Khelasi (Pengikat Besi)
Apabila diberikan sebagai kombinasi dengan transfusi, terapi khelasi dapat menunda
onset dari kelainan jantung dan, pada beberapa pasien, bahkan dapat mencegah kelainan
jantung tersebut.
Chelating agent yang biasa dipakai adalah DFO yang merupakan kompleks
hidroksilamin dengan afinitas tinggi terhadap besi. Rute pemberiannya sangat penting
untuk mencapai tujuan terapi, yaitu untuk mencapai keseimbangan besi negatif (lebih
banyak diekskresi dibanding yang diserap). Karena DFO tidak diserap di usus, maka rute
pemberiannya harus melalui parenteral (intravena, intramuskular, atau subkutan).
Dosis total yang diberikan adalah 30-40mg/kg/hari diinfuskan selama 8-12 jam saat
pasien tidur selama 5 hari/minggu.
Transplantasi Sel Stem Hematopoetik (TSSH)
TSSH merupakan satu-satunya yang terapi kuratif untuk thalassemia yang saat ini
diketahui. Prognosis yang buruk pasca TSSH berhubungan dengan adanya hepatomegali, fibrosis
portal, dan terapi khelasi yang inefektif sebelum transplantasi dilakukan. Prognosis bagi
penderita yang memiliki ketiga karakteristik ini adalah 59%, sedangkan pada penderita yang
tidak memiliki ketiganya adalah 90%.Meskipun transfusi darah tidak diperlukan setelah
transplantasi sukses dilakukan, individu tertentu perlu terus mendapat terapi khelasi untuk
menghilangkan zat besi yang berlebihan. Waktu yang optimal untuk memulai pengobatan
tersebut adalah setahun setelah TSSH. Prognosis jangka panjang pasca transplantasi , termasuk
fertilitas, tidak diketahui. Biaya jangka panjang terapi standar diketahui lebih tinggi daripada
biaya transplantasi. Kemungkinan kanker setelah TSSH juga harus dipertimbangkan.

53
Terapi Bedah
Splenektomi merupakan prosedur pembedahan utama yang digunakan pada pasien
dengan thalassemia. Limpa diketahui mengandung sejumlah besar besi nontoksik(yaitu, fungsi
penyimpanan). Limpa juga meningkatkan perusakan sel darah merah dan distribusi besi. Fakta-
fakta ini harus selalu dipertimbangkan sebelum memutuskan melakukan splenektomi.. Limpa
berfungsi sebagai penyimpanan untuk besi nontoksik, sehingga melindungi seluruh tubuh dari
besi tersebut. Pengangkatan limpa yang terlalu dini dapat membahayakan.
Sebaliknya, splenektomi dibenarkan apabila limpa menjadi hiperaktif, menyebabkan
penghancuran sel darah merah yang berlebihan dan dengan demikian meningkatkan kebutuhan
transfusi darah, menghasilkan lebih banyak akumulasi besi.
Splenektomi dapat bermanfaat pada pasien yang membutuhkan lebih dari 200-250 mL /
kg PRC per tahun untuk mempertahankan tingkat Hb 10 gr / dL karena dapat menurunkan
kebutuhan sel darah merah sampai 30%.

54
Gambar 8. Splenektomi
Risiko yang terkait dengan splenektomi minimal, dan banyak prosedur sekarang
dilakukan dengan laparoskopi. Biasanya, prosedur ditunda bila memungkinkan sampai anak
berusia 4-5 tahun atau lebih. Pengobatan agresif dengan antibiotik harus selalu diberikan untuk
setiap keluhan demam sambil menunggu hasil kultur. Dosis rendah Aspirin® setiap hari juga
bermanfaat jika platelet meningkat menjadi lebih dari 600.000 / μL pasca splenektomi.
Diet
Pasien dianjurkan menjalani diet normal, dengan suplemen sebagai berikut : asam folat,
asam askorbat dosis rendah, dan alfa-tokoferol. Sebaiknya zat besi tidak diberikan, dan makanan
yang kaya akan zat besi juga dihindari. Kopi dan teh diketahui dapat membantu mengurangi
penyerapan zat besi di usus.

55
A,9th, thalassemia
Riwayat keluarga
Anemia Hemolitik
Kerja hati&limpa meningkat
Pucat
HepatosplenomegaliIcterik sklera
F. Skrining
Dapat dilakukan skrining premarital dengan menggunakan pedigree. Atau bisa juga dilakukan
pemeriksaan terhadap setiap wanita hamil berdasar ras, melalui ukuran eritrosit, kadar Hb A2
(meningkat pada thalassemia-β). Bila kadarnya normal, pasien dikirim ke pusat yang bisa
menganalisis rantai α.
G. Prognosis
Prognosis bergantung pada tipe dan tingkat keparahan dari thalassemia. Seperti dijelaskan sebelumnya, kondisi klinis penderita thalassemia sangat bervariasi dari ringan bahkan asimtomatik hingga berat dan mengancam jiwa.
VIII. Kerangka Konsep
IX. Kesimpulan
Anakperempuan, 9 tahun, dating ke RSMH dengan keluhan distensi abdomen mengalami anemia
hemolitik e.c thalasemia

56
Daftar Pustaka
1. Berhman, RE; Kliegman, RM ; Arvin: Nelson Ilmu Kesehatan Anak, volume 2, edisi 15.
Penerbit Buku Kedokteran EGC, Jakarta : 2005, hal1708-1712
2. Berhman, RE; Kliegman, RM and Jensen, HB: Nelson Text Book of Pediatrics, 16th
edition. WB Saunders company, Philadelphia: 2000, page 1630-1634
3. Permono, H. BAmbang; Sutaryo; Windiastuti, Endang; Abdulsalam, Maria; IDG
Ugrasena: Buku Ajar Hematologi-Onkologi Anak, Cetakan ketiga. Penerbit Badan
Penerbit IDAI, Jakarta : 2010, hlm 64-84
4. Paediatrica Indonesiana, The Indonesian Journal of pediatrics and Perinatal Medicine,
volume 46, No.5-6. Indonesian Pediatric Society, Jakarta: 2006, page 134-138
5. Ananta Yovita. Terapi Kelasi Pada Thalassemia . Sari Pustaka. 2009
6. Atmakusuma, Djumhana, dkk., 2009. Buku Ajar Ilmu Penyakit Dalam Jilid II. Edisi V.
Jakarta Pusat: Interna Publishing.
7. Bakta, I Made. 2009. Hematologi klinik ringkas. EGC : Jakarta
8. Darling D. THALASSEMIA. . United states of america www.daviddarling.info( akses 2
Desember 2007 )
9. Kartoyo, Purnamawati. Pengaruh Penimbunan Besi Terhadap Hati pada Thalassemia.
Sari Pediatri. 2003. 05(01): 34-38.