laporan pkl c-organik
TRANSCRIPT

LAPORAN
PELAKSANAAN PRAKTIK KERJA LAPANGAN
BALAI PENGKAJIAN TEKNOLOGI PERTANIAN (BPTP)
JAWA TENGAH
VALIDASI METODE PENETAPAN C-ORGANIK
DALAM PUPUK ORGANIK
Oleh:
Nama : Eka Rusadi
NIM : 4311413067
Program Studi : Kimia
JURUSAN KIMIA
FAKULTAS MATEMATIKA DAN ILMU PENGETAHUAN ALAM
UNIVERSITAS NEGERI SEMARANG
2016i

HALAMAN PENGESAHAN
Laporan hasil Praktik Kerja Lapangan di Balai Pengkajian Teknologi Pertanian Jawa
Tengah 01 Februari 2016 sampai 01 Maret 2016 telah diselesaikan dan disahkan untuk
memenuhi salah satu syarat perkuliahan di Fakultas Matematika dan Ilmu Pengetahuan Alam
(MIPA) Jurusan Kimia Universitas Negeri Semarang pada :
Hari :
Tanggal :
Dosen Pembimbing PKL Pembimbing Lapangan
Jurusan Kimia FMIPA Unnes BPTP Jawa Tengah
Nuni Widiarti, S.Pd, M.Si Endah Winarni, S.T
NIP.19781028 200604 2 001 NIP 19691102 199403 2 003
Mengetahui,
Ketua Jurusan Kimia FMIPA Kepala BPTP Jawa Tengah
Dr. Nanik Wijayanti, M. Si Dr. Ir. Moh. Ismail Wahab, M.Si
NIP. 19691023 199603 2 002 NIP. 19650617 199103 1 002
ii

KATA PENGANTAR
Puji syukur penulis panjatkan kehadirat Allah SWT, yang telah melimpahkan
Rahmat-Nya sehingga penulis mampu menyelesaikan penyusunan laporan Praktek Kerja
Lapangan ini sesuai dengan waktu yang direncanakan. Laporan Praktek Kerja Lapangan ini
disusun untuk melengkapi syarat dalam menyelesaikan mata kuliah Praktek Kerja Lapangan
(PKL) di Jurusan Kimia Fakultas Matematika dan Ilmu Pengetahuan Alam Universitas Negeri
Semarang.
Laporan ini merupakan hasil dari kegiatan Praktek Kerja Lapangan yang telah
dilaksanakan di Balai Pengkajian Teknologi Pertanian (BPTP) Jawa Tengah tanggal 01
Februari 2016 sampai 01 Februari 2016.
Laporan ini dapat terselesaikan tidak lepas dari bantuan semua pihak, maka pada
kesempatan ini penulis mengucapkan terima kasih pada:
1.Dr. Nanik Wijayanti, M. Si selaku Ketua Jurusan Kimia.
2.Dr. Jumaeri, M.Si selaku Koordinator Program Studi Kimia yang telah membantu dan
membimbing penulis dalam perizinan PKL.
3.Nuni Widiarti, S.Pd, M.Si selaku dosen pembimbing PKL yang telah memberikan
bimbingan dan pengarahan kepada penulis.
4.Dr. Ir. Moh Ismail Wahab, M.Si selaku Kepala Balai Pengkajian Teknologi Pertanian Jawa
Tengah.
5.Endah Winarni, S.T selaku pembimbing lapangan yang telah mengarahkan, mendampingi,
dan membimbing penulis dalam melaksanakan PKL di Balai Pengkajian Teknologi
Pertanian Jawa Tengah.
6.Para staf dan karyawan Balai Pengkajian Teknologi Pertanian Jawa Tengah, Ibu Yulis, Ibu
Indra, Ibu Yatmi, Bapak Sutrisno, Bapak Rifai, dan masih banyak lagi yang tidak dapat
disebutkan satu persatu, yang telah banyak membantu membimbing dan memberikan
informasi selama PKL.
7.Bapak, Ibu, dan keluarga tercinta yang telah memberikan dukungan baik moril maupun
materil.
8.Teman satu kelompok PKL Aden, Ayang, Lala, dan Titis atas kerjasama dan bantuan
selama PKL berlangsung.
iii

9.Seseorang yang selalu memberi semangat selama pelaksanaan PKL dan membantu dalam
penyelesaian laporan PKL ini.
10.Sahabat dan teman-teman satu angkatan Kimia 2013 yang tidak bisa saya sebutkan satu
per satu.
Penulis menyadari bahwa masih banyak kekurangan dalam laporan Praktik Kerja
Lapangan ini. Oleh karena itu, saran dan kritik yang membangun senantiasa penulis terima
agar laporan ini menjadi lebih baik dan bisa bermanfaat untuk kita semua.
Semarang, Februari 2016
Penulis
iv

ABSTRAK
VALIDASI METODE PENETAPAN C-ORGANIK
DALAM PUPUK ORGANIK
Eka Rusadi, Praktik Kerja Lapangan di Balai Pengkajian Teknologi Pertanian Jawa Tengah, Bukit Tegalepek Ungaran
Jurusan Kimia Fakultas Matematika dan Ilmu Pengetahuan Alam Universitas Negeri Semarang
Pupuk adalah substansi atau bahan yang mengandung satu atau lebih zat yang dibutuhkan untuk pertumbuhan dan perkembangan tanaman. Material pupuk dapat berupa bahan organik ataupun non-organik (mineral). Pupuk berbeda dari suplemen. Pupuk mengandung bahan baku yang diperlukan pertumbuhan dan perkembangan tanaman, sementara suplemen seperti hormon tumbuhan membantu kelancaran proses metabolisme. Menurut bahan bakunya pupuk diklasifikasikan menjadi dua jenis, yaitu pupuk anorganik (buatan) dan pupuk organik.
Penetapan kadar C-organik dalam contoh pupuk organik dilakukan untuk mengetahui jumlah kandungan C-organik dalam pupuk organik. Dasar pengujiannya, karbon sebagai senyawa organik akan mereduksi Cr6+ yang berwarna jingga menjadi Cr3+ yang berwarna hijau yang terbentuk setara dengan kadar karbon dan dapat diukur dengan spektrofotometer pada panjang gelombang 561 nm. Validasi metode merupakan salah satu upaya laboratorium untuk membuktikan bahwa suatu metode uji yang digunakan senantiasa memberikan hasil yang dapat dipertanggungjawabkan, benar dan dapat dipercaya. Dalam validasi metode uji, parameter-parameter unjuk kerja metode yang dievaluasi dalam percobaan ini yaitu linearitas, limit deteksi, presisi, dan akurasi.
Dari hasil pengujian validasi metode penetapan C-organik pada sampel pupuk organik diperoleh linearitas C-organik dengan nilai regresi y = 0,0016x + 0,0037 dengan nilai linearitas koefisien korelasi (r) sebesar 0,9993 dan dari analisis uji diperoleh kandungan C-organik sebesar 6.713%, uji limit deteksi sebesar 59,8897 mg/L, uji presisi melalui repitabilitas dengan nilai pressentase RSD sebesar 4,2565% dan uji akurasi melalui persentase recovery dengan nilai sebesar 103,75%. Serta estimasi ketidakpastian kadar air sebesar 1,268375%; massa contoh 0,004110 gram; volume labu takar 0,085020 mL; spektrofotometer 0,013294 Abs; recovery 0,06748; dan presisi metode 0,042565. Semua parameter validasi telah memenuhi syarat yang ditetapkan.
Kata Kunci: Pupuk, C-Organik, Spektrofotometer, Validasi Metode.
v

DAFTAR ISI
Halaman
HALAMAN JUDUL .......................................................................................... i
HALAMAN PENGESAHAN ............................................................................ ii
KATA PENGANTAR ....................................................................................... iii
ABSTRAK ......................................................................................................... v
DAFTAR ISI ...................................................................................................... vi
DAFTAR TABEL .............................................................................................. viii
DAFTAR GAMBAR ......................................................................................... ix
DAFTAR LAMPIRAN ...................................................................................... x
BAB I. PENDAHULUAN
A. Latar Belakang Masalah .................................................................. 1
B. Perumusan Masalah ......................................................................... 2
C. Tujuan ............................................................................................. 2
D. Tempat dan Waktu PKL .................................................................. 3
E. Metode Pelaksanaan PKL ............................................................... 3
F. Sistematika Laporan ........................................................................ 3
BAB II. TINJAUAN PUSTAKA
A. Gambaran Umum BPTP Jawa Tengah ............................................ 5
B. Pupuk ............................................................................................... 9
C. C-Organik ........................................................................................ 11
D. Spektrofotometer ............................................................................. 12
E. Validasi Metode Analisis ................................................................ 14
F. Estimasi Ketidakpastian Pengukuran .............................................. 17
G. Sumber-Sumber Ketidakpastian ...................................................... 18
H. Klasifikasi Komponen Ketidakpastian ............................................ 19
vi

BAB III. METODE PENELITIAN
A. Tempat dan Waktu ................................................................................. 21
B. Alat dan Bahan ....................................................................................... 21
C. Metode Percobaan .................................................................................. 22
D. Penetapan Kadar Air (Metode Gravimetri) ............................................ 25
E. Pengukuran ............................................................................................. 25
F. Perhitungan dan Olah Data .................................................................... 26
BAB IV. HASIL DAN PEMBAHASAN
A. Linieritas ................................................................................................ 29
B. Limit Deteksi .......................................................................................... 29
C. Presisi (Repitabilitas) ............................................................................. 30
D. Akurasi ................................................................................................... 31
E. Estimasi Ketidakpastian Pengukuran ..................................................... 31
BAB V. PENUTUP
A. Simpulan ................................................................................................ 37
B. Saran ....................................................................................................... 38
DAFTAR PUSTAKA ........................................................................................ 39
LAMPIRAN ....................................................................................................... 41
vii

DAFTAR TABEL
Halaman
Tabel.1 SDM BPTP Jateng Menurut Jenjang Fungsional .......................................... 7
Tabel.2 SDM BPTP Jateng Menurut Kelompok Golongan ....................................... 8
Tabel.3 SDM BPTP Jateng Menurut Jenjang Fungsional Peneliti ............................ 8
Tabel.4 SDM BPTP Jateng Menurut Jenjang Fungsional Penyuluh Pertanian ....... 8
Tabel.5 SDM BPTP Jateng Menurut Jenjang Fungsional Litkayasa ......................... 8
Tabel.6 Nilai ketidakpastian Baku Gabungan ............................................................ 36
Tabel.7 Linearitas C-Organik ..................................................................................... 51
Tabel.8 Data Hasil Perhitungan Limit Deteksi .......................................................... 48
Tabel.9 Data Repitabilitas C-Organik ........................................................................ 50
Tabel.10 Hasil Perhitungan Recovery C-Organik (%) ................................................. 52
Tabel.11 Penetapan Kadar Air dan Faktor Koreksi BKM ........................................... 55
Tabel.12 Sumber-sumber Ketidakpastian .................................................................... 57
Tabel.13 Sumber-sumber Ketidakpastian Kadar Air ................................................... 58
Tabel.14 Ringkasan Ketidakpastian Kadar Air ............................................................ 60
Tabel.15 Data Hasil Ringkasan Recovery .................................................................... 62
Tabel.16 Daftar Data Sumber Ketidakpastian x C3 ..................................................... 63
Tabel.17 Ringkasan Nilai Ketidakpastian Konsentrasi Spike Larutan Baku ............... 65
Tabel.18 Data Hasil Presisi Metode ............................................................................. 66
viii

DAFTAR GAMBAR
Halaman
Gambar.1 Kantor BPTP Jawa Tengah ...................................................................... 5
Gambar.2 Struktur Organisasi BPTP Jawa Tengah .................................................. 7
Gambar.3 Cause and Effect Diagram atau Fish Bone C-Organik ............................ 31
Gambar.4 Cause and Effect Diagram atau Fish Bone kadar air sampel ................... 32
Gambar.5 Diagram Alir Penetapan Kadar Air .......................................................... 41
Gambar.6 Diagram Alir Pembuatan Larutan Standar C-Organik ............................. 42
Gambar.7 Diagram Alir Pembuatan Larutan Sampel ............................................... 43
Gambar.8 Diagram Alir Pembuatan Larutan Spike 1000 mg/L ................................ 44
Gambar.9 Diagram Alir Pembuatan Larutan Sampel + Spike .................................. 45
Gambar.10 Grafik Linearitas C-Organik ..................................................................... 46
Gambar.11 Cause and Effect Diagram atau Fish Bone x C3 ..................................... 63
ix

DAFTAR LAMPIRAN
Halaman
Lampiran. 1 Diagram Alir Kerja ................................................................................... 41
Lampiran. 2 Data dan Contoh Perhitungan Lineritas C-Organik ................................. 46
Lampiran. 3 Data dan Contoh Perhitungan Limit Deteksi C-Organik ......................... 48
Lampiran. 4 Data dan Contoh Perhitungan Repitabilitas C-Organik ........................... 50
Lampiran. 5 Data dan Contoh Perhitungan Recovery C-Organik ................................. 52
Lampiran. 6 Data dan Contoh Perhitungan Kadar Air .................................................. 55
Lampiran. 7 Data dan Perhitungan Estimasi Ketidakpastian ........................................ 57
Lampiran. 8 Dokumentasi ............................................................................................. 69
x

1
BAB I
PENDAHULUAN
A. Latar Belakang
Pupuk organik sangat bermanfaat bagi peningkatan produksi pertanian baik kualitas
maupun kuantitaas, mengurangi pencemaran lingkungan, dan meningkatkan kualitas lahan
secara berkelanjutan. Penggunaaan pupuk organik dalam jangka panjang dapat
meningkatkan produktivitas lahan dan dapat mencegah degredasi lahan.
Sumber bahan untuk pupuk organik sangat beranekaragam, dengan karakteristik
fisik dan kandungan kimia yang sangat beragam sehingga pengaruh dari penggunaan
pupuk organik terhadap lahan dan tanaman dapat bervariasi. Selain itu, peranannya cukup
besar terhadap perbaikan sifat kimia biologi tanah serta lingkungan (Basriman. MP, 2011).
Di dalam pupuk organik terdapat berbagai macam bahan organik yang dapat
dimanfaatkan oleh tanaman untuk tumbuh dan berkembang. Akan tetapi tidak semua pupk
cocok untuk semua jenis tanaman. Kemungkinan ada juga bahan di dalam pupuk yang
dapat merusak atau bahkan mematikan suatu tanaman.
Oleh karena itu mengetahui kandungan bahan organik di dalam pupuk organik
adalah hal yang penting untuk mengetahui tindakan apa yang tepat untuk mengolah pupuk
organik tersebut. Peneitian ini bertujuan untuk mengetahui seberapa besar kandungan C-
organik yang terdapat di dalam pupuk organik. Pengujian kandungan C-organik dalam
pupuk organik dilakukan dengan cara yang mengacu pada Buku Petunjuk Teknis Analisis
Kimia Tanah, Tanaman, Air, dan Pupuk Organik. Namun dalam perkembangan metode
analisis tersebut telah dimodifikasi oleh laboratorium untuk menyesuaikan dengan kondisi
laboratorium. Untuk mengevaluasi metode tersebut perlu dilakukan validasi metode dan
estimasi ketidakpastian pengukuran.
Validasi metode adalah konfirmasi bahwa suatu metode dapat memenuhi
persyaratan tujuan penggunaannya, yaitu melalui uji petunjuk (unjuk) kerja metode yang
bersangkutan dan mengumpulkan bukti atau hasilnya. Validasi metode merupakan slah
satu upaya laboratorium untuk membuktikan bahwa suatu metode uji yang digunakan
senantiasa memberikan hasil yang dapat dipertanggungjawabkan, benar dan dapat
dipercaya. Dalam validasi metode uji, parameter-parameter unjuk kerja metode yang

2
dievaluasi dalam percobaan ini yaitu linieritas, limit deteksi, presisi, dan akurasi (Sumardi,
2002).
B. Perumusan Masalah
1. Bagaimana uji validasi penetapan kandungan C-organik dalam pupuk organik?
2. Bagaimana cara menentukan estimasi ketidakpastian pengukuran penetapan kandungan
C-organik di dalam pupuk organik?
C. Tujuan PKL
Tujuan dilakukannya validasi penetapan C-organik dalam pupuk organik adalah
untuk mengetahui kandungan C-organik dalam pupuk organik, untuk memvalidasi metode
penetapan C-organik, dan untuk menetapkan nilai ketidakpastian pengukuran metode
pengukuran penetapan C-organik dan pengoprasian instrumen spektrofotometer. PKL ini
diharapkan dapat memberi manfaat:
1. Bagi Peneliti
Mampu mengembangkan kemampuan dalam memvalidasi metode-metode uji,
khususnya pengukuran C-organik dalam pupuk organik.
2. Bagi Universitas Negeri Semarang
Memberikan pengalaman kerja langsung kepada para mahasiswa Universitas Negeri
Semarang, khususnya mahasiswa Kimia Universitas Negeri Semarang.
3. Bagi Instansi (BPTP Jawa Tengah)
Mampu meningkatkan kepercayaan lebih dari instansi-instansi luar terhadap uji-uji
yang dilakuakan di BPTP Jawa Tengah dengan tervalidasinya metode uji.
D. Tempat dan Waktu PKL
Kegiatan PKL bertempat di laboratorium Balai Pengkaji Teknologi Pertanian
(BPTP) Jawa Tengah yang berlokasi di Bukit Tegalepek Ungaran. Kegiatan PKL ini
dilaksanakan pada tanggal 01 Ferbuari 2016 sampai dengan 01 Maret 2016.
E. Metode Pelaksanaan PKL
Pelaksanaan PKL di Laboratorium BPTP Jawa Tengah, antara lain:
1. Pengambilan Sampel

3
Sampel yang diuji adalah pupuk organik yang diambil dari arsip BPTP yang berlokasi
di bukit Tegalepek Ungaran.
2. Preparasi
Preparasi dilakukan pada tanggal 05 sampai 09 februari 2016 meliputi persiapan
sampel, penipangan sampel, pembuatan larutan standar C-organik, pembuatan
Recovery larutan.
3. Penetapan
Penetapan uji dilakukan pada tanggal 10 sampai 13 Februari 2016 meliputi penetapan
kadar C-organik pupuk organik, dan pengukuran absorbansi larutan menggunakan
instrumen spektrofotometer merk spektronik 21D.
4. Analisis Data
Analisis data dilakukan setelah semua pengukuran dilaksanakan dengan menggunakan
program statistika.
5. Studi Pustaka
Studi pustaka dilakukan untuk mendapatkan keterangan dan informasi mengenai proses
penentuan kandungan C-organik dalam pupuk organik. Studi pustaka diperoleh dari
literatur-literatur yang terdapat di BPTP Jawa Tengah serta buku-buku yang
berhubungan dengan penentuan kandungan C-organik daam pupuk organik.
F. Sistematika Laporan
Laporan Praktik Kerja Lapangan ini tersusun dalam sistematika penyusunan sebagai
berikut:
BAB I PENDAHULUAN
Bagian pendahuluan memuat Latar Belakang, Perumusan Masalah, Tujuan dan Manfaat,
Tempat dan Waktu Pelaksanaan, Metode Pelaksanaan dan Sistematika penyusunan
laporan.
BAB II TINJAUAN PUSTAKA
Bab ini berisi tentang gambaran umum perusahaan dan tinjauan-tinjauan pustaka atau
landasan teori yang berhubungan dengan kegiatan Praktik Kerja Lapangan.
BAB III METODE PENELITIAN

4
Bab ini berisi tentang subyek penelitian, metode pengumpulan data, prosedur penelitian
dan metode analisis data.
BAB IV HASIL DAN PEMBAHASAN
Bagian ini berisi tentang hasil dan pembahasan dari kegiatan Praktik Kerja Lapangan yang
telah dilakukan.
BAB V PENUTUP
Bagian ini berisi tentang simpulan dan saran.

5
BAB II
TINJAUAN PUSTAKA
A. Gambaran Umum BPTP Jawa Tengah
Gambar 1. Kantor BPTP Jawa Tengah
1. Sejarah BPTP
BPTP Jawa Tengah dahulu bernama BPTP Ungaran dibentuk berdasarkan
Surat Keputusan Menteri Pertanian No. 798/Kpts/OT.210/1994 tanggal 13 Desember
1994. BPTP Ungaran merupakan gabungan (merger) dari Balai Informasi Pertanian
Ungaran, Balai Informasi Pertanian Yogyakarta, Sub Balai Penelitian Ternak Klepu,
Sub Balai Penelitian Perikanan Laut Semarang, Kebun Percobaan Muktiharjo, Kebun
Percobaan Ngemplak, Kebun Percobaan Batang, Stasiun Penelitian Tanah dan
Laboratorium Hortikultura Yogyakarta serta Proyek Penelitian Penyelamatan Hutan
Tanah dan Air (P3HTA) Badan Litbang Pertanian. Wilayah kerja BPTP Ungaran
meliputi Provinsi Jawa Tengah dan Daerah Istimewa Yogyakarta. Bersamaan dengan
kebijakan nasional tentang otonomi daerah, BPTP Ungaran berubah nama menjadi
BPTP Jawa Tengah yang ditetapkan dengan Surat Keputusan Mentan Pertanian
No.350/Kpts/OT. 210/6/2001 tanggal 14 Juni 2001. Implikasi dan perubahan tersebut
antara lain adalah wilayah kerja BPTP Jawa Tengah menjadi hanya Provinsi Jawa
Tengah. Sedangkan unit-unit yang berada di DIY menjadi BPTP DIY.
Bersamaan dengan kebijakan nasional tentang otonomi daerah, BPTP Ungaran
berubah nama menjadi BPTP Jawa Tengah yang ditetapkan dengan Surat Keputusan
Menteri Pertanian No.350/Kpts/OT.210/6/2001 tanggal 14 Juni 2001. Implikasi dan
perubahan tersebbut antara lain adalah wilayah kerja BPTP Jawa Tengah menjadi
hanya di Provinsi Jawa Tengah.

6
2. Tugas Pokok dan Fungsi BPTP Jawa Tengah
Berdasar Peraturan Menteri Pertanian Nomor: 16/permentan/OT.140/3/2006
a. Tugas Pokok
Melaksanakan pengkajian, perakitan dan pengembangan teknologi pertanian
tepat guna spesifik lokasi.
b. Fungsi
1) Pelaksanaan inventarisasi dan identifikasi kebutuhan teknologi pertanian tepat
guna spesifik lokasi.
2) Pelasanaan penelitian, pengkajian dan perakitan teknologi pertanian tepat guna
spesifik lokasi.
3) Pelaksanaan pengembangan teknologi dan diseminasi hasil pengkajian serta
perakitan materi penyuluhan.
4) Penyiapan kerjasama, informasi, dokumentasi, serta penyebarluasan dan
pendayagunaan hasil pengkajian, perakitan dan pengembangan teknologi
pertanian tepat guna spesifik lokasi.
5) Pemberian pelayanan teknik kegiatan pengkajian, perakitan dan
pengembangan teknologi pertanian tepat guna spesifik lokasi.
6) Melaksanakan urusan tata usaha dan rumah tangga balai.
3. Program BPTP Jawa Tengah
1. Inventarisasi, pengelolaan dan pengembangan sumberdaya pertanian spesifik
lokasi.
2. Pengkajian teknologi inovatif spesifik lokasi dan agribisnis unggulan daerah.
3. Pengkajian komunikasi, diseminasi dan penjaringan umpan balik teknologi
pertanian spesifik lokasi.
4. Pengembangan model agribisnis berbasis inovasi pertanian.
5. Penelitian dan pengkajian berbasis kemitraan dan keperluan pembangunan
pertanian spesifik lokasi berdasar permintaan.
6. Analisis dan sintesa kebijakan pembangunan pertanian daerah.
7. Pengembangan kapasitas kelembagaan litbang pertanian.
8. Pengembangan sumberdaya informasi, komunikasi, diseminasi dan penjaringan
umpan balik IPTEK.

7
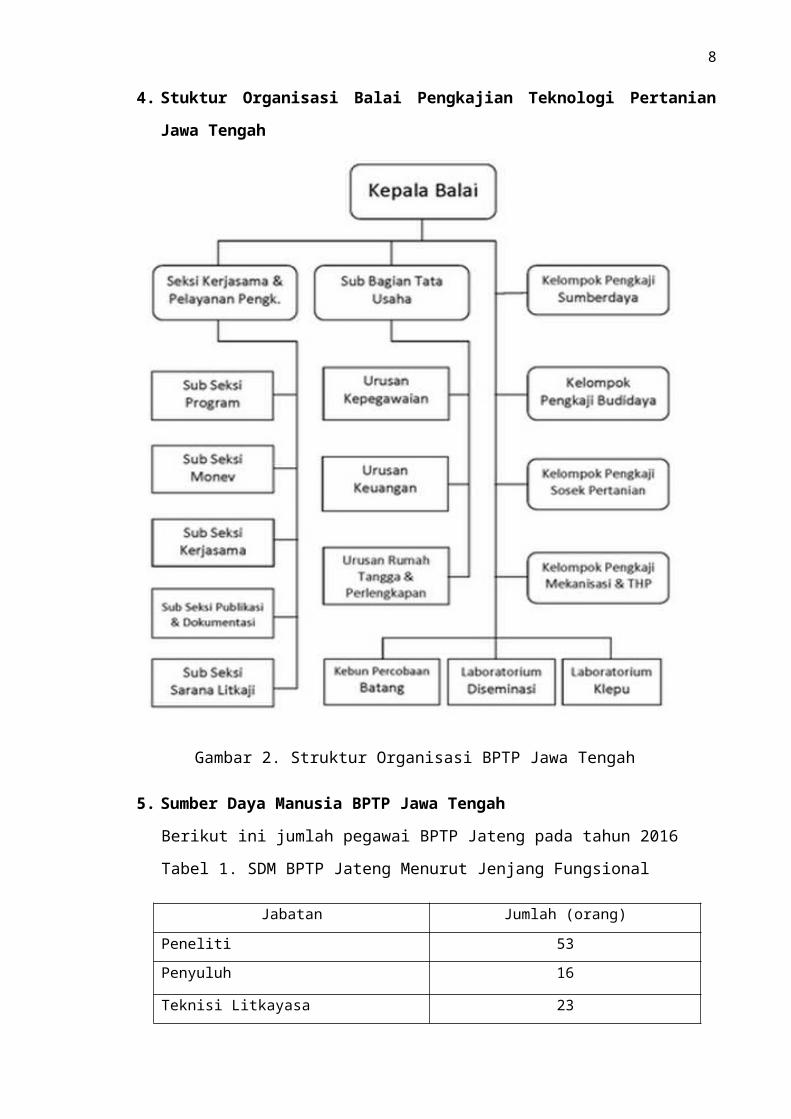
4. Stuktur Organisasi Balai Pengkajian Teknologi Pertanian Jawa Tengah
Gambar 2. Struktur Organisasi BPTP Jawa Tengah
5. Sumber Daya Manusia BPTP Jawa Tengah
Berikut ini jumlah pegawai BPTP Jateng pada tahun 2016
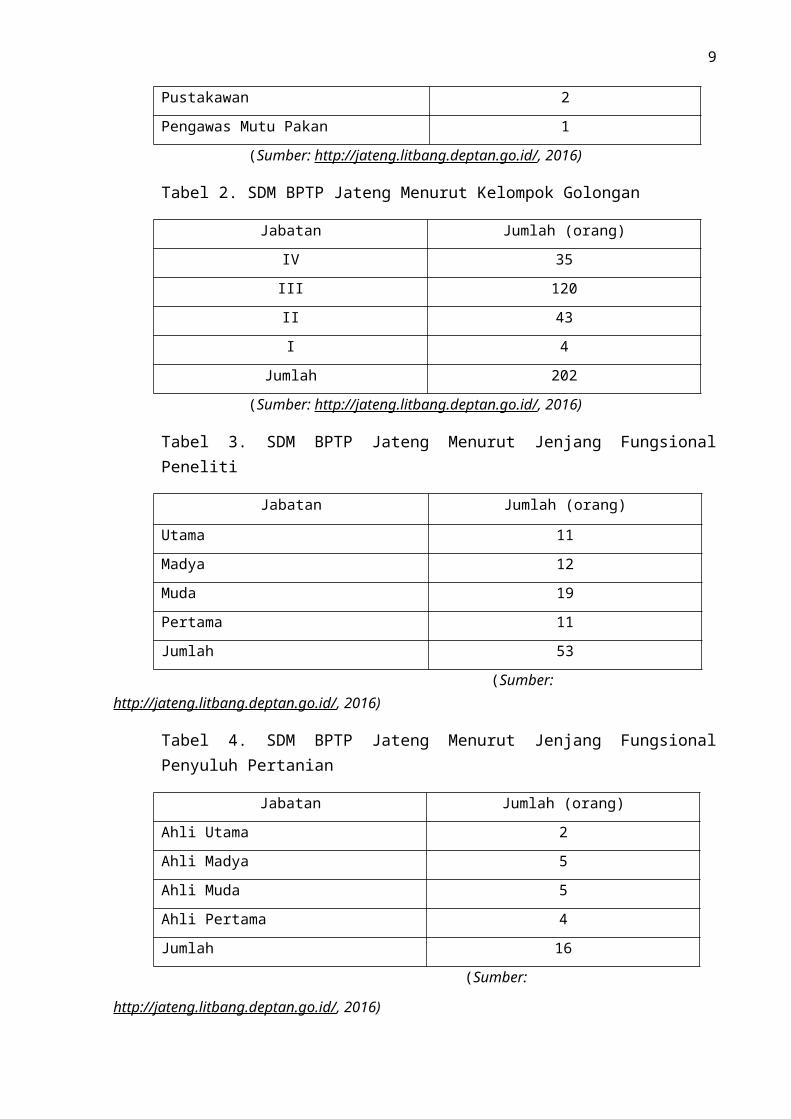
Tabel 1. SDM BPTP Jateng Menurut Jenjang Fungsional
Jabatan Jumlah (orang)
Peneliti 53
Penyuluh 16
Teknisi Litkayasa 23
Pustakawan 2
Pengawas Mutu Pakan 1
(Sumber: http://jateng.litbang.deptan.go.id/, 2016)

8
Tabel 2. SDM BPTP Jateng Menurut Kelompok Golongan
Jabatan Jumlah (orang)
IV 35
III 120
II 43
I 4
Jumlah 202
(Sumber: http://jateng.litbang.deptan.go.id/, 2016)
Tabel 3. SDM BPTP Jateng Menurut Jenjang Fungsional Peneliti
Jabatan Jumlah (orang)
Utama 11
Madya 12
Muda 19
Pertama 11
Jumlah 53
(Sumber: http://jateng.litbang.deptan.go.id/, 2016)
Tabel 4. SDM BPTP Jateng Menurut Jenjang Fungsional Penyuluh Pertanian
Jabatan Jumlah (orang)
Ahli Utama 2
Ahli Madya 5
Ahli Muda 5
Ahli Pertama 4
Jumlah 16
(Sumber: http://jateng.litbang.deptan.go.id/, 2016)
Tabel 5. SDM BPTP Jateng Menurut Jenjang Fungsional Litkayasa
Jabatan Jumlah (orang)
Litkayasa Penyedia 5
Litkayasa Pelaksana Lanjutan 10
Litkayasa Pelaksana 7
Litkayasa Pemula 1
Jumlah 23
(Sumber: http://jateng.litbang.deptan.go.id/, 2016)

9
B. Pupuk
Pupuk adalah material yang ditambahkan pada media tanam atau tanaman untuk
mencukupi kebutuhan hara yang diperlukan tanaman sehingga mampu berproduksi
dengan baik. Material pupuk dapat berupa bahan organik ataupun non-organik (mineral).
Pupuk berbeda dari suplemen, pupuk mengandung bahan baku yang diperlukan
pertumbuhan dan perkembangan tanaman, sementara suplemen seperti hormon tumbuhan
membantu kelancaran proses metabolisme. Meskipun demikian ke dalam pupuk,
khususnya pupuk buatan dapat ditambahkan sejumlah material suplemen.
Menurut bahan bakunya pupuk diklasifikasikan menjadi dua jenis, yaitu pupuk
anorganik (buatan) dan pupuk organik.
1. Pupuk Anorganik
Pupuk anorganik adalah pupuk yang dibuat oleh pabrik-pabrik pupuk dengan
meramu bahan-bahan kimia anorganik berkadar hara tinggi. Misalnya urea berkadar N
45-46% (setiap 100 kg urea terdapat 45-46 kg hara nitrogen) (Lingga dan Marsono,
2000).
Pupuk anorganik atau pupuk buatan dapat dibedakan menjadi pupuk tunggal dan
pupuk majemuk. Pupuk tunggal adalah pupuk yang hanya mengandung satu unsur
hara misalnya pupuk N, pupuk P, pupuk K, dan sebagainya. Pupuk majemuk adlah
pupuk yangmengansung lebih dari satu unsur hara misalnya N + P, P + K, N + K, N +
P + K, dan sebagainya (Hardjowigeno, 2004).
Ada beberapa keuntungan dari pupuk anorganik, yaitu:
a. Pemberiannya dapat terukur dengan tepat;
b. Kebutuhan tanaman akan hara dapat dipenuhi dengan perbandingan yang tepat;
c. Pupuk anorganik tersedia dalam jumlah cukup; dan
d. Pupuk anorganik mudah diangkat karena jumlahnya relatif sedikit dibandingkan
dengan pupuk organik. Pupuk anorganik mempunyai kelemahan, yaitu selain
hanya mempunyai unsur makro, pupuk anorganik ini sangat sedikit ataupun
hampir tak mengandung unsur hara mikro (Lingga dan Marsono, 2000).
2. Pupuk organik
Pupuk organik adalah pupuk yang tersusun dari materia makhluk hidup, seperti
pelapukan sisa-sisa tanaman, hewan, dan manusia. Pupuk organik dapat berbentuk
padat atau cair yang digunakan untuk memperbaiki sifat fisik, kimia, dan biologi

10
tanah. Pupuk organik mengandung banyak bahan organik daripada kadar haranya.
Sumber bahan organik dapat berupa kompos, pupuk hijau, pupuk kandang, sisa panen
(jerami, brangkasan, tongkol jagung, bagas tebu, dan sabut kelapa), limbah ternak,
limbah industri yang menggunakan bahan pertanian, dan limbah kota (sampah).
Berbagai hasil penelitian mengindikasikan bahwa sebagian besar lahan
pertanaian intensif menurun produktivitasnya dan telah mengalami degradasi lahan,
terutama terkait dengan sangat rendahnya kandunan karbon organik dalam tanah, yaitu
2%. Psdahal untuk memperoleh produktivitas optimal dibutuhkan karbon organik
sekitar 2,5% (Basriman. MP, 2011).
Pupuk oraganik sangat bermanfaat bagi peningkatan produksi pertanian baik
kualitas maupun kuantitas, mengurangi pencemaran lingkungan, dan meningkatkan
kualitas lahan secara berkelanjutan. Penggunaan pupuk organik dalam jangka panjang
dapat meningkatkan produktivitas lahan dan dapat mencegah degradasi lahan
(Basiman. MP, 2011).
Sumber bahan untuk pupuk organik sangat beranekaragam, dengan
karakteristik fisik dan kandungan kimia yang snagt beragam sehingga pengaruh dari
penggunaan pupuk organik terhadap lahan dan tanaman dapat bervariasi. Selain itu,
peranannya cukup besar terhadap perbaikan sifat fisik, kimia, kimia, iologi tanah, serta
lingkungan (Basriman. MP, 2011).
Pupuk organik yang ditambahkan ke dalam tanah akan mengalami beberapa
kalli fase perombakan oleh mikroorganisme tanah untuk menjadi humus. Bahan
organik juga berperan sebagai sumber energi dan makanan mikroba tanah sehinggga
dapat meningkatkan aktivitas mikroba tersebut dalam penyediaan hara tanaman.
Penambahan bahan organik disamping sebagai hara bagi tanaman, juga sebagai
sumber energi dan hara bagi mikroba (Basriman. MP, 2011).
Bahan dasar pupuk organik yang berasal dari sisa tanaman sedikit mengandung
bahan berbahaya. Penggunaan pupuk kandang, limbah industri dan limbah kota
sebagai bahan dasar kompos berbahaya karena banyak mengandung logam berat dan
asam-asam organik yang dapat mencemari lingkungan. Selama proses pengomposan,
beberapa bahan berbahaya ini akan terkonsentrasi dalam produk akhir pupuk
(Basriman. MP, 2011).
Untuk itu diperlukan seleksi bahan dasar kompos yang mengandung bahan-bahan berbahaya dan beracun (B3). Pupuk organik dapat berperan sebagai pengikat butiran primer menjadi butir sekunder tanah dalam pembentukan pupuk. Keadaan ini

11
mempengaruhi penyimpanan, penyediaan air, aerasi tanah, dan suhu tanah. Bahan organik dengan karbon dan nitrogen yang banyak, seperti jerami atau sekam lebih besar pengaruhnya.pasda perbaikan sifat-sifat fisik tanah dibanding dengan bahan organik yang terdekomposisi seperti kompos (Basriman. MP, 2011).
Menurut Basriman (2011) pupuk organik memiliki fungsi kimia yang sangat penting seperti:
1. Penyediaan hara makro (nitrogen, fosfor, kalium, kalsium, magnesium, dan sulfur)
dan mikro seperti zink, tembaga, kobalt, barium, mangan, dan besi, meskipun
jumlahnya relatif sedikit;
2. Meningkatkan kapasitas tukar kation (KTK) tanah;
3. Membentuk senyawa kompleks dengan ion logam yang meracuni tanaman seperti
magnesium, besi, dan mangan.
C. C-Organik
C-organik penting untuk mikroorganisme tidak hanya sebagai unsur hara tetapi
juga sebagai pengkondisi sifat fisik tanah yang memperngaruhi karakteristik agregat
dan air tanah. Seringkali ada hubungan langsung antara persentase C-organik total dan
karbon daRi biomassa mikroba yang ditemukan dalam tanah pada zona iklim yang
sama. C-organik juga berhubungan dengan aktivitas enzim tanah. Di perkebunan teh
Gembung, C-organik tanah juga digunakan untuk menetukan dosis asam-asam organik
dan apabila ditambahkan ke dalam tanah akan meningkatkan kandungan senyawa
organik dan tanah yang dicirikan dengan meningkatnya kadar C-organik tanah.
Tanaman mengambil unsur karbon berupa CO2 dari udara bebas (atmosfer). Kegiatan
ini dilakukan oleh organ tanaman yang memiliki klorofil, umumnya bagian tanaman
yang berwarna hijau dan erdapat di atas tanah. Klorofil mampu menyerap energi
cahaya (terutama sinar matahari) dan mengubahnya menjadi energi kimia. Energi
tersebut digunakan untuk menghasilkan CO2 menjadi senyawa organik termasuk
karbohidrat (Fauzi, 2008).
Menurut Fauzi (2008), kadar CO2 dalam atmosfer relatif stabil yakni 0,03%
volume atau 0,05 mg/L udara. Tanpa adanya CO2 di udara, maka kehidupan tanaman
akan terhenti. Jika kehidupan tanaman terhenti, maka kehidupan makhluk lain
termasuk manusia dan hewan juga akan terhenti.
Afandi (2002) menyebutkan bahwa sumber utama CO2 di alam berasal dari
dekomposisi bahan organik berupa sisa-sisa tanaman ataupun hewan dan dari respirasi
invertebrata, abakteri, serta fungi. Berdasarkan perhitungan Unspenkii cit, jumlah CO2
12
yang dihasilkan oleh penapasan populasi heterotrof per tahun diperkirakan sebagai
berikut:
Binatang invertebrata : 3,7 x 109 ton
Bakteri : 51,4 x 109 ton
Fungi/jamur : 8,8 x 109 ton
Akar tanaman :71,5 x 109 ton
Jumlah CO2 : 135,4 x 109 ton
Keperluan seluruh tanaman yang hidup diperkirakan 80 x 109 ton karbon per
tahun. Dengan persediaan CO2 dalam udara sebesar 0,03% volume, maka CO2 tersebut
akan habis diserap tanamn dalam waktu beberapa dekade saja. Berkat adanya daur
(siklus) yang menghasilkan CO2 maka kadar gas tersebut relatif stabil (Afandi, 2002).
Karbon penting sebagai bahan pembangun bahan organik, karena sebagian
besar bahan kering tanaman terdiri dari bahan organik, sumber karbon dapat dikatakan
banyak, dalam ruangan tertutup yang berisi CO2, fotosintesa tetap aktif (Fauzi, 2008).
D. Spektrofotometer
Spektrofotometer merupakan alat yang digunakan untuk mengukur absorbansi dengan
cara melewatkan cahaya dengan panjang gelombang tertentu pada suatu obyek kaca atau
kuarsa yanng disebut kuvet. Sebagian dari cahaya tersebut akan diserap dan sisanya akan
dilewatkan. Nilai aabsorbansi dari cahaya yang dilewatkan akan sebanding dengan
konsentrasi larutan di dalam kuvet (Day & Underwood, 1994).
Spektrofotometer sangat berhubungan dengan pengukuran jauhnya pengabsorbansian
energi cahaya oleh suatu sistem kimia sebagai fungsi panjang gelombang dengan
absorbansi maksimum dari suatu sistem kimia sebagai fungsi panjang gelombang dengan
absorben maksimum dari suatu unsur atau senyawa. Konsenrasi unsur atau senyawa dapat
dihitung dengan menggunakan kurva standar yang diiukur pada panjang gelombang
absorban tersebut, yaitu panjang gelombang yang diperoleh dari hasil nilai absorbansi
yang tertinggi (Day & Underwood, 1994).
Spektrum absorben selain tergantung pada sifat unsur kimia, juga bergantung pada
faktor-faktor lain. Perubahan pelarut sering menghasilkan pergeseran dari pita absorbansi.
Larutan pembanding dalam spektrofotometri pada umumnya adalah pelarut murni atau
suatu larutan blanko yang mengandung sedikit zat yang akan ditetapkan atau tidak sama
sekali (Day & Underwood, 1994).
Secara garis besar spektrofotometer terdiri dari 4 bagian penting yaitu:
13
1. Sumber cahaya, pada spektrofotometer sumber cahaya haruslah memiliki pancaran
radiasi yang stabil dan intensitas tinggi.
2. Monokromator adalah alat yang berfungsi untuk menguraikan cahaya polikromatis
menjadi beberapa komponen panjang gelombang tertentu.
3. Kuvet spektrofotometer adalah suatu alat yang digunakan sebagai tempat contoh atau
cuplikan yang akan dianalisis
4. Detektor, peranan detektor penerima adalah memberikan respon terhadap cahaya pada
berbagai panjang gelombang. Detektor akan mengubah cahaya menjadi sinyal listrik
yang selanjutnya akan ditampilkan oleh penampil data dalam bentuk jarum penunjuk
atau angka digital.
Menurut Ghozali (2010) dalam Ardana (2012), dalam analisis menggunakan
spektrofotometer dikenal adanya trasmitansi dan absorbansi, adapaun hubungan dari
keduanya adalah sebagai berikut:
1. Transmitansi
Apabila suatu berkas sinar radiasi dengan intensitas Io dilewatkan melalui suatu
larutan dalam wadah transparan maka sebagian radiasi akan diserap sehingga
intensitas radiasi yang diteruskan It menjadi lebih kecil dari Io.
Transmitansi dengan simbol T dari larutan merupakan fraksi dari radiasi yang
diteruskan atau ditransmisikan oleh larutan, yaitu:
T = ¿Io Transmitansi biasanya dinyatakan dalam persen (%).
2. Absorbansi
Absorbansi dengan simbol A dari suatu larutan merupakan logaritma dari 1/T atau
logaritma Io/It.
A = log 1T atau A = - log T
Absorbansi berbanding langsung dengan tebal larutan dan konsentrasi larutan (hukum
Beer), yaitu: A = a.b.C
Harga a tergantung pada satuan yang digunakan untuk b dan c. Apabila konsentrasi
dinyatakan dalam mol/liter dan tebal sel dalam cm, maka absorbtivitas disebut
absorbtivitas molar dan diberi simbol ԑ. Jadi, persamaannya dapat ditulis yaitu:
A = ԑ.b.c (ԑ merupakan satuan Lcm-1mol-1)
Harga absorbansi (A), absorbtivitas molar (ԑ), tebal larutan (b) dalam cm dan
konsentrasi larutan (c) dalam mol/L.
Menurut Hendayana (1994), beberapa persyaratan yang harus diperhadikan
supaya Hukum Lambert-Beer dapat dipakai, yaitu:

14
a. Konsentrasi harus rendah;
b. Zat yang diukur harus stabil;
c. Cahaya yang dipakai harus monokromatik;
d. Larutan yang diukur harus jernih.
E. Validasi Metode Analisis
Seperti yang tertuang dalam ISO/IEC, validasi diartikan sebagai kegiatan
konfirmasi melalui pengujian dan pengadaan bukti yang objektif bahwa persyaratan
tertentu untuk suatu maksut harus dipenuhi.
Validasi metode analisis adalah suatu proses penilaian terhadap metode analisis
tertentu berdasarkan percobaan laboratorium untuk membuktikan bahwa metode tersebut
memenuhi persyaratan untuk digunakan (Harmita, 2004). Selain itu, validasi metode
dilakukan jika terjadi perubahan kondisi antara kondisi analisis dan kondisi pada saat
validasi metode, atau terjadi perubahan metode dari metode standar. Beberapa manfaat
validasi metode metode analisis adalah untuk mengevaluasi unjuk kerja suatu metode
analisis, menjamin prosedur analisis, menjamin keakuratan dan kedapatulangan hasil
prosedur analisis, dan mengurangi resiko penyimpangan yang mungkin timbul
(Wulandari, 2007).
Validasi metode bertujuan untuk mengetahui sejauh mana penyimpangan yang
tidak dapat dihindari dari suatu metode pada kondisi normal dimana seluruh elemen
terkait telah dilaksanakan dengan baik dan benar. Penggunaan metode pengujian yang
benar sangat diperlukan untuk mengetahui tingkat akurasi dan presisi dari suatu data hasil
pengujian. Sebagai konsekuensinya, laboratorium harus memvalidasi metode pengujian
sebelum metode ini digunakan. Laboratorium harus memvalidasi metode tidak baku,
metode yang didesain atau dikembangkan oleh laboratorium, dan modifikasi dari metode
baku untuk mengkonfirmasi bahwa metode itu sesuai untuk penggunaan yang
dimaksudkan. Secara sederhana hasil uji yang abash dapat digambarkan sebagai hasil uji
yang mempunyai akurasi dan presisi yang baik (Sumardi, 2002).
Dalam proses validasi metode, parameter-parameter unjuk kerja metode ditentukan
dengan menggunakan peralatan yang memenuhi spesifikasi, bekerja dengan baik dan
terkalibrasi secara memadai. Secara umum, validasi metode mancakup penentuan yang
berkaitan dengan alat dan metode (Wulandari, 2007).
Prosedur analisis yang harus divalidasi meliputi beberapa jenis pengujian, yaitu
adanya pengotor, uji limit untuk mengendalikan keberadaan pengotor, serta uji kuantitatif

15
komponen aktif atau komponen lain. Selain itu, terdapat beberapa parameter dalam
validasi analisis, dimana pemilihan parameter yang akan diuji tergantung dari jenis dan
metode pengujian yang akan divalidasi.
Dalam validasi metode ini, parameter-parameter unjuk kerja metode yang
dievaluasi dalam percobaan ini, yaitu:
1. Linieritas
2. Limit deteksi
3. Presisi (repitabilitas)
4. Akurasi (Recovery).
1. Lineritas
Linieritas adalah kemampuan metode analisis yang memberikan respon yang
secara langsung atau dengan bantuan transformasi matematik yang baik, proporsional
terhadap konsentrasi analit dalam sampel dalam rentang yang ditentukan. Lineritas
suatu metode harus diuji untuk membuktikan adanya hubungan yang linear antara
konsentrasi analit dan respon alat. Hubungan lineritas dinyatakan dalam koefisien
korelasi (r). Dalam suatu analis, harga koefisien korelasi (r) ini sebaiknya >0.99 (Miller
dan Miller, 1991).
r=ƩXY −ƩXY
n
√ Ʃ X2−(ƩX )2
n √ƩY 2−(ƩY )2
n
Keterangan:
n : Jumlah ulangan
X : Konsentrasi
Y : Absorbans
2. Limit deteksi
Limit deteksi dari suatu metode analisis adalah nilai parameter uji batas, yaitu
konsentrasi analit terendah yang dapat dideteksi, tetapi tidak dikuantitasi pada kondisi
percobaan yang dilakukan. Limit deteksi dinyatakan dalam konsentrasi terendah analit
(persen, bagian per milyar) dari suatu contoh yang masih dapat dideteksi oleh alat
(Sumardi, 2002). Limit kuantitasi dari suatu metode analisis adalah nilai parameter
penentuan kuantitatif senyawa yang terdapat dalam konsentrasi rendah dalam matriks.
Limit kuantitasi adalah konsentrasi analit terendah dalam sampel yang dapat ditentukan
dengan presisi dan akurasi yang dapat diterima pada kondisi eksperimen yang

16
ditentukan. Limit kuantitasi dinyatakan dalam konsentrasi analit (persen, bagian per
milyar) dalam sampel.
Menurut Harmita (2004), penentuan limit deteksi instrument (LDI) dapat dihitung
berdasarkan pada standar deviasi (SD) respon dan kemiringan (slope) linieritas baku
dengan rumus:
LDI = Nilai rata-rata konsentrasi terkecil + 3 SD
SD = Standar Deviasi
3. Presisi (repitabilitas)
Cisca (2009), keseksamaan menyatakan seberapa dekat suatu hasil pengukuran
satu dengan yang lainnya. Semakin dekat nilai-nilai hasil pengulangan pengukuran
maka semakin presisi pengukuran tersebut.
Presisi menggambarkan kesalahan acak dari suatu hasil pengukuran. Kesalahan
acak berasal dari pengaruh-pengaruh yang tidak dapat diperkirakan, bervariasi terhadap
ruang, dan bersifat sementara. Kesalahan acak sulit untuk dihindari, banyak
berhubungan dengan instrumen ukur, peralatan contoh yang diukur, prosedur, dan
lingkungan (Harmita, 2004).
Penentuan presisi dapat dilakukan dengan uji repitabilitas, presisi antara, dan
reprodusibillitas. Repitabilitas atau keterulangan dilakukan dalam kondisi yang sama
dalam interval waktu yang singkat. Kondisi sama ini dapat diartikan dengan
penggunaan laboratorium yang sama, analisis yang sama, dan pereaksi serta peralatan
yang sama. Presisi antara menyatakan variasi dalam laboratorium yang sama dengan
melakukan pengujian pada hari yang berbeda, oleh analis yang berbeda, dan
menggunakan pereaksi serta peralatan yang berbeda. Reprodusibiltas menyatakan
presisi antar laboratorium sehingga dilakukan pada kondisi yang telah ditentukan di
laboratorium yang berbeda, pada hari yang berbeda, dan menggunakan peralatan serta
pereaksi yang berbeda pula (Sumardi, 2002).
Menurut Harmita (2004), presisi antara menyatakan variasi dalam laboratorium
yang sama dengan melakukan pengujian pada hati yang berbeda. Reprodusibilitas
menyatakan presisi antar laboratorium sehingga dilakukan pada kondisi yang telah
ditentukan laboratorium yang berbeda, pada hari yang berbeda dan menggunakan
peralatan serta pereaksi yang berbeda pula. Presisi dinyatakan sebagai presentase
Relative Standar Deviation (% RSD) dari suatu seri pengukuran.
SD = √∑ (xi−x )2
n−1

17
% RSD = SDx x 100%
Keterangan:
xi : nilai data pengukuran
x : rata-rata pengukuran
n : jumlah ulangan
RSD menunjukkan ketelitian dari metode uji:
RSD ≤ 1 % : sangat teliti
1% < RSD ≤ 2% : teliti
2% < RSD ≤ 5% : ketelitian sedang
RSD > 5% : tidak teliti
4. Akurasi (Recovery)
Akurasi menunjukkan kedekatan pengukuran terhadap nilai sebenarnya. Akurasi
menyatakan kesesuaian antara hasil dengan nilai sebenarnya. Akurasi menggambarkan
systematic error. Sumber kesalahan dapat berasal dari kelembaban, bahan referensi,
ketidakpastian yang diberikan sertifikat, metode analisis, dan lain-lain
.
Suatu metode dikatakan valid jika nilai presentasi Recovery suatu standar antara
90-110% dengan perhitungan sebagai berikut (Sumardi, 2002)
% Recovery=C 2−C 1C 3
x 100 %
Keterangan:
C1 = Konsentrasi contoh tanpa analit (mg/L)
C2 = Konsentrasi contoh yang ditambah analit (mg/L)
C3 = Konsentrasi analit (spike) (mg/L)
F. Estimasi Ketidakpastian Pengukuran
Menurut SNI 19-17025-2000 (2000), laboratorium pengujian harus
mempunyai dan menerapkan prosedur untuk mengestimasi ketidakpastian pengukuran.
Estimasi yang wajar harus berdasarkan pada pengetahuan atas unjuk kerja metode pada
lingkup pengukuran dan harus menggunaka, sebagai contoh, pengalaman sebelumnya dan
data validasi.
Pengukuran bertujuan untuk menentukan besaran nilai yang diukur. Pada
umumnya hasil pengukuran hanya merupakan nilai dugaan terhadap nilai benar besaran

18
yang diukur. Nilai dugaan mengandung banyak faktor kesalahan atau ketidakpastian yang
mempengaruhi hasil pengukuran. Oleh karena itu, indikator pengukur kualitas hasil
pengukuran diperlukan untuk mengetahui sumber - sumber yang mempengaruhi hasil
pengukuran dan memenuhi persyaratan yaitu universal, konsisten, dapat diukur, dan dapat
ditransfer dari suatu pengukuran ke pengukuran yang lain. Indikator tersebut dikenal
dengan nama ketidakpastian. Oleh karena itu, hasil pengukuran hanya lengkap apabila
disertai dengan nilai ketidakpastian pengukurannya (Sumardi, 2002).
Menurut Sumardi (2002), ketidakpastian didefinisikan sebagai suatu parameter
yang menetapkan rentang nilai dugaan yang didalamnya diperkirakan terletak nilai benar
berada. Ketidakpastian ditunjukkan dengan tanda (±) yang dihubungkan dengan hasil
pengukuran yang mencirikan dispersi (penyebaran) nilai untuk dicantumkan dalam nilai
yang diukur. Konsep ketidakpastian berdasarkan pada besaran teramati yang diperoleh
dengan pengukuran. Hal ini berbeda dengan konsep ideal kesalahan yang berdasarkan
pada besaran yang tidak dapat diketahui. Estimasi ketidakpastian pengukuran dilakukan
apabila pengujian memberikan hasil numerik (kuantitatif), sedangkan untuk pengujian
yang bersifat kualitatif (misalnya: lulus/tidak, positif/negatif) atau berdasarkan pada visual
atau dapat diraba (tactile) tidak dipersyaratkan untuk mengestimasi ketidakpastian
pengukurannya.
Ketidakpastian dan kesalahan mempunyai kaitan satu sama lain.
Ketidakpastian menggunakan semua kesalahan yang diketahui menjadi suatu rentang
tunggal. Ketidakpastian berupa rentang atau kisaran dan tidak perlu nilai benar, sedangkan
kesalahan berupa pengamatan tunggal dan perlu nilai benar (Sumardi, 2002).
G. Sumber - Sumber Ketidakpastian
Dalam praktik, terdapat berbagai kemungkinan sumber ketidakpastian
pengukuran seperti definisi besaran ukur yang tidak lengkap, realisasi definisi besaran
ukur yang tidak sempurna, pengambilan sampel yang tidak mewakili keseluruhan besaran
ukur yang didefinisikan, pengetahuan yang tidak memadai tentang pengaruh kondisi
lingkungan terhadap proses pengukuran atau pengukuran kondisi lingkungan yang tidak
sempurna, bisa personal dalam membaca peralatan analog, resolusi atau diskriminasi
peralatan, nilai yang diberikan pada standar pengukuran atau bahan acuan, nilai konstanta
dan parameter lain yang diperoleh dari sumber luar dan digunakan dalam algoritma
reduksi data, pendekatan dan asumsi yang tercakup dalam metode dan prosedur
pengukuran, dan variasi pengamatan berulang terhadap besaran ukur dalam kondisi yang
tampak sama (Sumardi, 2002).

19
Interpretasi dari sumber ketidakpastian pengukuran dalam aplikasinya untuk
proses pengujian dapat mencakup tetapi tidak terbatas pada pengambilan sampel yang
tidak representatif, ketidakhomogenan asal sampel, kontaminasi selama pengambilan dan
penyiapan sampel, kemurnian pereaksi dan larutan, pengaruh dan interferensi matriks dan
koreksi blank (Komite Akreditasi Nasional, 2003).
H. Klasifikasi Komponen Ketidakpastian
Menurut Komite Akreditasi Nasional (2003), ketidakpastian pengukuran terdiri
dari beberapa komponen yang dapat diklasifikasikan menurut metode yang digunakan
untuk menaksir nilai numeriknya, yaitu:
1. Komponen tipe A yang bersumber pada kesalahan acak dari hasil pengukuran
berulang dan dinyatakan dalam bentuk SD, kemudian dievaluasi dengan analisis
statistika dari serangkaian pengamatan. Evaluasi tipe A dapat diterapkan untuk
mengestimasi ketidakpastian dari efek personal (pengulangan penimbangan,
pemipetan, titrasi, pengujian, pengukuran dengan alat, dan sebagainya), kinerja alat
(presisi hasil pengukuran), dan kinerja metode (presisi hasil uji menggunakan metode
terkait).
2. Komponen tipe B yang bersumber pada pengalaman atau informasi yang tersedia
(berasal dari kesalahan acak dan sistematik) dan dinyatakan dalam bentuk SD,
kemudian dievaluasi dengan analisis nonstatistika dari serangkaian pengamatan.
Evaluasi tipe B dapat digunakan untuk mengestimasi ketidakpastian baku, misalnya
nilai acuan dari standar, alat ukur gelas, variasi suhu ruang pengujian, dan hasil
kalibrasi alat pengukuran. Proses evaluasi tipe B menggunakan data atau informasi
sertifikasi kalibrasi alat, sertifikasi bahan acuan, spesifikasi pabrik, data dari
handbook, data dari katalog, dan data dari manual alat yang berupa Quoted
Uncertainty (QU) yang diubah menjadi µ yang dihitung dari nilai QU yaitu dengan
membaginya dengan faktor pencakupan (k). Nilai k bergantung pada asumsi
probabilitas ketidakpastian tersebut. Ketidakpastian baku tipe ini berdasarkan atas
sebaran atau distribusi kejadiannya. QU memiliki salah satu dari distribusi:
a. Distribusi Normal
Sebaran nilai ukur yang berada di sekitar suatu harga dan sebaran nilai ukurnya
dibatasi oleh µc ± k (bergantung pada tingkat kepercayaan yang dipilih). Bila tingkat
kepercayaan:
68 % maka faktor k = 1
90 % maka faktor k = 1,6
95 % maka faktor k = 1,96 = 2

20
99 % maka faktor k = 2,6
99,73 % maka faktor k = 3
Untuk menghitung ketidakpastian distribusi digunakan rumus:
µ = QUk
b. Distribusi Rektanguler (Segi Empat)
Distribusi ini dipakai jika kita yakin bahwa kesalahan yang lebih besar lebih
mungkin terjadi. Untuk menghitung ketidakpastian dari distribusi ini digunakan rumus
sebagai berikut:
µ = QU√3
c. Distribusi Trianguler (Segi Tiga)
Distribusi ini dipakai jika kita yakin bahwa kesalahan yang lebih kecil lebih
mungkin terjadi. Untuk menghitung ketidakpastian dari distribusi ini digunakan rumus
sebagai berikut:
µ = QU√6
Untuk evaluasi ketidakpastian tipe B, apabila tidak ada informasi tambahan
mengenai spesifikasi suatu alat atau bahan, maka diasumsikan distribusinya segi
empat. Komponen ketidakpastian masing-masing diestimasi sehingga ekuivalen
dengan SD. Komponen ini disebut sebagai ketidakpastian baku (µ) (Sumardi, 2002).

21
BAB III
METODE PENELITIAN
Percobaan ini bertujuan untuk memvalidasi dan menetapkan nilai ketidakpastian
pengukuran metode penetapan C-organik yang terkandung di dalam pupuk organik
menggunakan instrumen Spektronik 21D sehingga metode ini valid digunakan sebagai
analisis rutin di laboratorium kimia Balai Pengkajian Teknologi Pertanian Jawa Tengah.
A. Tempat dan Waktu
Praktik Kerja Lapangan (PKL) ini dilaksanakan mulai tanggal 01 Februari 2016
sampai dengan 01 Maret 2016 di laboratorium Balai Pengkajian Teknologi Pertanian (BPTP)
Jawa Tengah yang berlokasi di Bukit Tegalepek Sidomulyo Ungaran, Semarang.
B. Alat dan Bahan
1. Alat
Alat-alat yang digunakan yaitu spektronik 21D merk Milton Roy, neraca analitik,
cawan porselin, cawan porselin, oven listrik mek Memmert, desikator, labu takar volume
100 mL, dispenser skala 20 mL, pipet ukur 1, 2, 5, dan 10 mL.
2. Bahan
Bahan yang digunakan dalam PKL ini meliputi bahan uji dan bahan pereaksi. Bahan
uji yang diguanakan sebagai contoh dalam percobaan ini adalah conth pupuk organik
yang diambil dari arsip di BPTP Jawa Tengah yang berlokasi di Bukit Tegalepek
Ungaran dengan kode PO-02. Bahan pereaksi yang digunakan yaitu larutan standar C-
Organik 5000 mg/L, H2SO4 pa. 98%, BJ 1,84, K2Cr2O7 1 N, dan air bebas ion.

22
C. Metode Percobaan
Percobaan yang dilakukan terdiri dari preparasi, pengukuran, dan pengolahan
data. Dalam tahap preparasi dilakukan persiapan contoh, pembuatan larutan standar,
dan penetapan kadar air (metode gravimetri). Tahap berikutnya adalah pengukuran
contoh dan deret standar dengan menggunakan Spektronik 21D pada panjang
gelombang 561 nm nm. Parameter validasi meliputi linieritas, limit deteksi, presisi
melalui repitabilitas, dan akurasi melalui uji perolehan kembali (Recovery). Nilai
ketidakpastian pengukuran diperoleh dengan memperhitungkan berbagai sumber
ketidakpastian. Estimasi ketidakpastian pengukurannya terdiri dari penetapan kadar air
(metode gravimetri), massa contoh, volume labu takar, faktor pengenceran,
konsentrasi contoh, konsentrasi baku, Recovery, dan presisi metode. Berdasarkan hasil
percobaan diolah dengan menggunakan teknik statistika.
1. Persiapan Sampel / Contoh
a. Pencatatan Contoh
Contoh dari lapangan yang disertai dengan surat permintaan analisis yang
berisi daftar contoh dan jenis analisis yang diperlukan, diterima oleh
administrasi laboratorium. Dalam buku administrasi dicatat nomor permintaan
analisis, jumlah dan nomor contoh. Untuk setiap contoh dibuat nomor
laboratorium yang ditulis pula pada label karton. Administrasi laboratorium
juga membuat hasil analisis yang telah selesai dikerjakan. Surat permintaan
dan daftar hasil analisis didokumentasikan.
b. Menyebarkan contoh di atas tampah yang dialasi kertas sampul kemudian label
karton yang berisi nomor laboratorium contoh diselipkan di bawah kertas.
Kemudian bongkahan pupuk organik yang besar dikecilkan dengan tangan.
Sampel disimpan pada rak di ruangan khusus bebeas konaminan yang
terlindungi dari sinar matahari atau sampel dimasukkan ke dalam oven dengan
suhu 40°C.
2. Cara Kerja Penetapan Kadar C-Organik
a. Dasar penetapan
Karbon sebagai senyawa organik akan mereduksi Cr6+ yang berwarna jingga
menjadi Cr3+ yang berwarna hijau dalam suasana asam. Inensitas warna hijau
yang terbentuk setara dengan kadar karbon dan dapat diukur dengan
spektrofotometer pada panjang gelombang 561 nm.
Berikut adalah reaksi yang terjadi:

23
C-organik + 2K2Cr2O7 + 8H2SO4 2Cr(SO4)3 + 2K2SO4 + 8H2O + 3CO2
b. Persiapan Contoh
1) Menimbang secara teliti 0,050 gram contoh pupuk organik yang telah
halus ke dalam labu takar volume 100 mL. Menambahkan 5 mL larutan
K2Cr2O7 1 N (dikocok) dan 7 mL H2SO4 pekat (dikocok), membiarkan
larutan samapai dingin dan jika perlu sekali-kali dikocok. Mengencerkan
larutan sampai tanda tera setelah larutan dingin dengan air bebas ion,
kocok bolak-balik hingga homogen dan larutan diidamkan selama
semalam. Mengukur larutan dengan spektrofotometer pada panjang
gelombang 561 nm esok harinya (dilakukan sepuluh kali pengulangan).
2) Pembuatan larutan dikromat (K2Cr2O7) 1 N
Melarutkan 98,1 gram kalium dikromat dengan 600 mL air bebas ion
dalam gelas piala, dan menambahkan 100 mL asam sulfat pekat, kemudian
memanaskan larutan hingga larut sempurna, mengencerkan larutan dalam
labu takar 1 liter dengan air bebas ion sampai tanda batas setelah larutan
dingin.
c. Pembuatan larutan standar
1) Standar 0 ppm C
Memipet 5 mL larutan K2Cr2O7 1 N dan menambahkan 7 mL H2SO4 pa.
98% (dikocok) membiarkan larutan sampai dingin jika perlu sekali-kali
dikocok. Kemudian larutan diencerkan dengan air bebas ion dan setelah
dingin volume ditepatkan hingga tanda tera 100 mL, kocok bolak-balik
hingga homogen dan biarkan semalam.
2) Standar 50 ppm C
Memipet 1 mL larutan standar baku 5000 ppm C dimasukkan ke dalam
labu takar volume 100 mL, menambahkan 5 mL larutan K2Cr2O7 1 N dan
menambahkan 7 mL H2SO4 pa. 98% (dikocok) membiarkan larutan sampai
dingin jika perlu sekali-kali dikocok. Kemudian larutan diencerkan dengan
air bebas ion dan setelah dingin volume ditepatkan hingga tanda tera 100
mL, kocok bolak-balik hingga homogen dan biarkan semalam.
3) Standar 100 ppm C
Memipet 2 mL larutan standar baku 5000 ppm C dimasukkan ke dalam
labu takar volume 100 mL, menambahkan 5 mL larutan K2Cr2O7 1 N dan
menambahkan 7 mL H2SO4 pa. 98% (dikocok) membiarkan larutan sampai

24
dingin jika perlu sekali-kali dikocok. Kemudian larutan diencerkan dengan
air bebas ion dan setelah dingin volume ditepatkan hingga tanda tera 100
mL, kocok bolak-balik hingga homogen dan biarkan semalam.
4) Standar 150 ppm C
Memipet 3 mL larutan standar baku 5000 ppm C dimasukkan ke dalam
labu takar volume 100 mL, menambahkan 5 mL larutan K2Cr2O7 1 N dan
menambahkan 7 mL H2SO4 pa. 98% (dikocok) membiarkan larutan sampai
dingin jika perlu sekali-kali dikocok. Kemudian larutan diencerkan dengan
air bebas ion dan setelah dingin volume ditepatkan hingga tanda tera 100
mL, kocok bolak-balik hingga homogen dan biarkan semalam.
5) Standar 200 ppm C
Memipet 4 mL larutan standar baku 5000 ppm C dimasukkan ke dalam
labu takar volume 100 mL, menambahkan 5 mL larutan K2Cr2O7 1 N dan
menambahkan 7 mL H2SO4 pa. 98% (dikocok) membiarkan larutan sampai
dingin jika perlu sekali-kali dikocok. Kemudian larutan diencerkan dengan
air bebas ion dan setelah dingin volume ditepatkan hingga tanda tera 100
mL, kocok bolak-balik hingga homogen dan biarkan semalam.
6) Standar 250 ppm C
Memipet 5 mL larutan standar baku 5000 ppm C dimasukkan ke dalam
labu takar volume 100 mL, menambahkan 5 mL larutan K2Cr2O7 1 N dan
menambahkan 7 mL H2SO4 pa. 98% (dikocok) membiarkan larutan sampai
dingin jika perlu sekali-kali dikocok. Kemudian larutan diencerkan dengan
air bebas ion dan setelah dingin volume ditepatkan hingga tanda tera 100
mL, kocok bolak-balik hingga homogen dan biarkan semalam.
Esok harinya standar diukur dengan spektrofotometer pada panjang
gelombang 561 nm (dilakukan sepuluh kali pengulangan). Pengukuran ini
menghasilkan hubungan antara konsentrasi dengan absorbansi dalam
bentuk kurva kalibrasi standar C.
d. Pembuaatan spike 1000 ppm
Memipet larutan standar baku 5000 ppm C sebanyak 10 mL ke dalam labu
takar volume 50 mL, kemudian menghimpitkan larutan sampai tanda batas
dengan akuades, lalu dihomogenkan (1000 ppm).
e. Preparasi contoh + spike (C2)

25
Menimbang secara teliti 0,050 gram contoh pupuk organik yang telah halus ke
dalam labu takar volume 100 mL. Menambahkan 2 mL spike, kemudian
berturut-turut menambahkan 5 mL laruutan K2Cr2O7 1 N, dikocok, dan 7 mL
H2SO4 pa. 98% , dikocok lagi, membiarkan larutan sampai dingin jikaa perlu
sekali-kali dikocok. Setelah dingin volume ditepatkan hingga tanda tera 100
mL dengan air bebas ion, kemudian larutan dihomogenkan. Esoknya mengukur
absorbansi larutan dengan menggunakan spektrofotometer pada panjang
gelombang 561 nm (dilakukan sepuluh kali pengulangan).
D. Penetapan Kadar Air (Metode Gravimetri)
1. Dasar penetapan
Air dalam sampel tanah diuapkan dengan cara pengeringan oven pada suhu
105 ºC selama 3 jam untuk menghilangkan kadar air. Kadar air dari contoh
diketahui dari perbedaan bobot contoh sebelum dan setelah dikeringkan. Faktor
koreksi kelembapan dihitung dari kadar air contoh.
2. Cara Kerja
Timbang teliti 10 g contoh dalam cawan porselin bertutup yang sudah
diketahui bobotnya. Masukan ke dalam oven dan dikeringkam selama 3 jam pada
suhu 105 oC. Dinginkan dalam desikator dan timbang. Kemudian masukkan ke
dalam oven dan dikeringkan selama 30 menit dan didinginkan lalu timbang hingga
mendapatkan berat yang konstan.
Perhitungan
Kadar air (%) = (M 2−M 3)(M 2−M 1) x 100
Dimana:
M1 = Bobot cawan kosong (gram)
M2 = Bobot cawan + sampel sebelum dioven (gram)
M3= Bobot cawan + sampel setelah dioven (gram)
fk (faktor koreksi kadar air) = 100/(100 - % kadar air)
(dihitung dari kadar air contoh tanah dan digunakan sebagai faktor koreksi dalam perhitungan hasil analisis selain kadar air dan bahan ikutan)
.
E. Pengukuran

26
Larutan standar dan larutan berisi sampel diukur dengan Spektronik 21D dengan
panjang gelombang 561 nm.
F. Perhitungan dan Olah Data
Berdasarkan data hasil analisis yang diperoleh, lalu diolah secara statistika dengan
menggunakan persamaan-persamaan di bawah ini, yaitu:
a. Linieritas
Linieritas ditentukan dengan cara mengukur deret standar campuran.
Berdasarkan data yang diperoleh dibuat kurva kalibrasi standar C-organik. Dari
persamaan garis regresi yang diperoleh dari masing-masing kurva dapat ditentukan
koefisien korelasi (r) dengan rumus :
a = [∑ Y −(b∑ X )]/ n
b = ∑ XY −[ (∑ XY ) /n ]∑ x2−[∑ x2 /n ]
r = ∑ XY −[ (∑ XY )
n ]∑ x2−
(∑ x2)n √∑ y2−
(∑ y2 )n
Keterangan:
a = intersep X = konsentrasi
b = slope Y = absorbansi
n = jumlah pengulangan
b. Limit Deteksi dan Limit Kuantitasi
Limit deteksi ditentukan dengan cara mengukur konsentrasi standar
campuran terkecil sebanyak sepuluh kali pengulangan. Berdasarkan data yang
diperoleh maka ditentukan Limit Deteksi Instrumen (LDI) dengan rumus :
LDI = A standar terendah + 3 SD
LoQ = A standar terendah + 10 SD
Keterangan:
A : Absorbansi
SD : Standard Deviation (standar deviasi)
c. Presisi (Repitabilitas)

27
Repitabilitas ditentukan dengan pengulangan perlakuan contoh sebanyak
sepuluh kali pengulangan. Berdasarkan data yang diperoleh dihitung rata-rata (x),
standar deviasi (SD), dan % RSD.
x = ∑ xin
SD = √∑ (xi−x )2
n−1
% RSD = SDx x 100%
Keterangan :
xi = nilai data pengukuran
x = rata-rata pengukuran
n = jumlah ulangan
d. Akurasi
Penetapan akurasi dilakukan melalui uji perolehan kembali (Recovery) dengan
rumus:
% Recovery = C 2−C 1
C 3 x 100%
Keterangan:
C1 = Konsentrasi larutan sampel + spike
C2 = Konsentrasi laarutan sampel
C3 = Konsentrasi larutan spike
e. Estimasi Ketidakpastian Pengukuran
Pengolahan data dilakukan berdasarkan teknik statistika yaitu dengan
menentukan rata-rata (x) dan standar deviasi (SD). Setelah itu, dilanjutkan dengan
perhitungan estimasi ketidakpastian. Hal ini dilakukan untuk menentukan nilai
ketidakpastian dari suatu percobaan.
Tahapan estimasi ketidakpastian adalah sebagai berikut :
1) Menetukan spesifikasi yang diukur dengan formula / persamaan
2) Mengidentifikasi sumber ketidakpastian
a) Membuat daftar dari semua sumber ketidakpastian
b) Membuat daftar cause and effect diagram atau fish bone
3) Ketidakpastian baku (µ)

28
Mengkuantisasikan masing-masing komponen ketidakpastian, yaitu
nilai yang dikontribusikan oleh masing-masing sumber ketidakpastian dengan
tipe A atau tipe B.
a) Tipe A (dengan perhitungan statistika)
SD = µ
b) Tipe B (dengan cara nonstatistika)
i. Distribusi Normal
µ = QU
k
Nilai faktor cakupan (k) tergantung pada tingkat kepercayaannya.
ii. Distribusi rektangular (segi empat)
µ = QU√3
iii. Disttribusi triangular (segi tiga)
µ = QU√6
Keterangan:
QU = Quoted Uncertainty
k = faktor cakupan
4) Ketidakpastian gabungan (µc)
Ada tiga aturan gabungan, yaitu:
a) Aturan 1
Penjumlahan
Y = a + b + c +...
Ketidakpastian gabungannya
µc (Y) = √ ((µ(a))2+(µ(b))2+(µ(c))2+…)b) Aturan 2
Perkalian atau pembagian
Y = a.b.c atau Y= ab/c
Ketidakpastian gabungannya
µc (Y) = Y √ {( µ (a ) /a )2+(µ (b ) /b )2+( µ (c ) /c )2+…}c) Aturan 3
Pangkat
Y = an

29
Ketidakpastian gabungannya
µc (Y) = [nY µ (a ) ] /a5) Ketidakpastian diperluas/expanded uncertainty (U)
U = ketidakpastian gabungan x k
U = µc (Y) x k
Nilai k = 1.96 atau 2 apabila tingkat kepercayaan 95 %.
BAB IV
HASIL DAN PEMBAHASAN
Berdasarkan hasil percobaan dan pengolahan data maka diperoleh hasil
validasi dan estimasi ketidakpastian metode penetapan C-organik dalam contoh pupuk
organik di daerah BPTP Jawa Tengah yang berlokasi di Bukit Tegalepek Ungaran
dengan instrumen spektronik 21D. Parameter validasi yang diuji meliputi linieritas,
limit deteksi, presisi melalui repitibilitas, dan akurasi melalui uji perolehan kembali
(Recovery). Nilai pelaporan hasil uji disertai dengan ketidakpastian pengukurannya.
A. Linieritas
Uji linieritas suatu metode bertujuan membuktikan adanya hubungan yang
linier antara konsentrasi analit yang sebenarnya dengan respon alat. Parameter
menunjukkan adanya hubungan yang linier antara absorbansi dengan konsentrasi
analit dinyatakan dalam koefisien korelasi (r). Uji linieritas dari pengukuran deret
standar diperoleh data dan kurva kalibrasi standar C yang terdapat dalam lampiran 2.
Berdasarkan hasil analisis yang terdapat pada Gambar 8, diperoleh nilai regresi
y = 0,0016x + 0,0037 dan nilai koefisien korelasi (r) sebesar 0,9993. Didapatkan nilai
r = 0,9993, kurva berbentuk garis linier yang terdiri dari enam titik pengukuran yaitu
konsentrasi 0 mg/L, 50 mg/L, 100 mg/L, 150 mg/L, 200 mg/L, 250 mg/L. Penggunaan

30
enam konsentrasi cukup mewakili dalam proses validasi, karena pada cara kerja yang
digunakan dalam menetapkan kadar C-organik menggunakan enam standar
konsentrasi. Nilai r = 0,9993 yang diperoleh telah memenuhi syarat yang ditetapkan,
dengan ketentuan r = 0,99.
B. Limit Deteksi
Limit deteksi dilakukan dengan cara mengukur larutan standar C-organik terkecil
yaitu 50mg/L. Pengukuran dilakukan sebanyak sepuluh kali pengulangan. Data yang
dihasilkan ditentukan standar deviasinya. Karena pada konsentrasi terendah (0 mg/L) hasil
pengukuran menunjukkan absorbansi yang bernilai nol, maka penghitungan limit deteksi
dilakukan pada konsentrasi 50 mg/L. Hasil pengukuran limit deteksi instrumen dapat dillihat
pada Tabel 8, beserta contoh penghitungannya yang terdapat pada lampiran 3.
Parameter limit deteksi instrumen menunjukkan konsenrasi terkecil yang dapat
terbaca karena sinyal pengganggu dari instrumen. Pada konsentrasi terkecil alat sangat
terbatas dalam membedakan sinyal. Hasil pengukuran pada Tabel 8 diperoleh nilai
limit deteksi yang merupakan penjumlahan antara nilai rata-rat konsentrasi terkecil
ditambah dengan hasil perkalian tiga kali standar deviasi, pada pengukuran
konsentrasi terendah adalah 51,375 mg/L maka niali limit deteksinya sebesar 59,8897
mg/L. Berdasarkan nilai tersebut dapat diketahui bahwa penggunaan instrumen pada
penetapan C-organik dalam contoh pupuk organik dengan konsentrasi ≥ 59,8897 mg/L
dapat dipercaya sebagai sinyal alat terhadap analit, tetapi untuk konsentrasi analit <
59,8897 mg/L sinyal yang dihasilkan tidak dipercaya analit, melainkan noise. Nilai
59,8897 mg/L merupakan konsentrasi terendah yang masih dapat dipercaya pada
pengukuran dengan menggunakan instrumen tersebut.
C. Presisi
Uji presisi dilakukan melaui uji repirepettibilitas untuk variabilitas dan yang
dihasilkan dari suatu pengujian yang dilakukan pada kondisi yang sama. Presisi hasil
pengukuran digambarkan dalam bentuk persentase Relative Standard Deviation
(%RSD). Berdasarkan hasil pengukuran contoh pupuk organik dengan sepuluh kali
pengulangan diperoleh data seperti terddapat pada Tabel 9 dalam lampiran 4.

31
Pada Tabel 9 diperoleh nilai presentase RSD C-Organik sebesar 4,2565%.
Nilai yag diperoleh memenuhi syarat yang ditetapkan, yaitu 2% < RSD ≤ 5%
ketelitian sedang. Nilai persentase RSD yang diperoleh tersebut dikategorikan ke
dalam tingkat keterulangan hasil pengukuran yang baik dan termasuk ke dalam
pengukuran ketelitian yang sedang sehingga nilai dari kesalahan acak dari metode
tersebut kecil, sehingga hasil uji memberikan presisi yang baik. Kesalahan pada
penetapan persentase RSD bisa diakibatkan oleh ketidakpastian alat atau instrumen
yang digunakan, selain itu juga dapat berasal dari kesalahan pembacaan skala oleh
praktikan.
D. Akurasi
Penetapan akurasi dilakukan untuk mengetahui keakuratan suatu metode oleh
karena itu, dilakukan evaluasi akurasi metode melalui uji perolehan kembali
(Recovery). Nilai persentase Recovery yang mendekati 100% menunjukkan bahawa
metode tersebut mempunyai ketepatan yang baik dalam menunjukkan tingkat
kesesuaian nilai rata-rata dari suatu pengukuran yang sebanding dengan nilai
sebenarnya.
Akurasi dapat dilihat dari nilai Recovery spike yaitu dengan cara
menambahkan sejumlah analit (standar) ke dalam contoh yang telah diketahui
konsentrasinya. Nilai %Recovery beserta contoh perhitungan dapat dilihat pada Tabel
10 dalam lampiran 5.
Pada Tabel 10 lampiran 5 diperoleh nilai rata-rata persentase Recovery untuk
spike C-organik yang ditambahkan sebesar 103,75%. Nilai kisaran persentase
Recovery yang baik disyaratkan berada pada rentang 90-110% untuk konsentrasi
rendah. Nilai Recovery yang diperoleh untuk spike C-organik memenuhi persyaratan
maka metode ini dapat dikatakan akurat.
E. Estimasi Ketidakpastian Pengukuran

Recovery Mc
Kal
KA VLT
Kal
ET
Karbon Organik
Kurva KalibrasiPM
Oven
Massa C3
32
Evaluasi dari setiap sumber ketidakpastiannya adalah sebagai berikut:
Gambar 3. Cause and effect diagram atau fish bone C-organik

Massa Teruapkan
Kadar Air(%)
Massa Basah Oven
M2
M2
Ka
Ka
M3
Ka
KaM1 Ka
ET
33
Gambar 4. Cause and effect diagram atau fish bone kadar air sampel
Keterangan:
Ka : Kalibrasi
PM : Presisi Metode
KA : Kadar Air
VLT : Volume Labu Takar
ET : Efek Temperatur
IS : Instrumen Spektrofotometer
C3 : Konsentrasi spike
Evaluasi dari setiap sumber ketidakpastian adalah sebagai berikut:
1. Perhitungan ketidakpastian baku dari penetapan C-organik

34
Ketidakpastian baku dari lima sumber ketidakpastian dapat dikuantifikasi sebagai
berikut:
a. Ketidakpastian baku kadar air (metode gravimetri) (µc KA)
Ketidakpastian baku kadar air bersumber dari massa teruapkan, massa basah,
oven, homogenitas, dan repetibilitas. Diperoleh sebesar 1,268375%.
1) Ketidakpastian baku dari masing-masing massa teruapkan dan massa basah
diperoleh dari kalibrasi, sensitivitas, dan repetibilitas.
a) Kalibrasi timbangan yang diterbitkan oleh laboratorium kalibrasi yang
telah terakreditasi, tertulis perbedaan antara berat yang sebenarnya
dengan berat yang terbaca pada skala adalah ± 0,00582 gram dengan
tingkat kepercayaan 95% dengan faktor cakupan dua. Evaluasi
ketidakpastian berdasarkan tipe B dengan asumsi distribusi normal. Nilai
ketidakpastian bakunya yaitu 0,00291 gram. Ketidakpastian kalibrasi
harus dihitung dua kali penimbangan contoh. Nilai ketidakpastian baku
dari massa basah yaitu 0,00411 gram.
b) Sensitivitas Timbangan diabaikan karena penimbangan berat dilakukan
pada timbangan yang sama dan perbedaan nilai yang kecil dari
penimbangan wadah sebelum dan sesudah ditambahkan contoh sehingga
sensitivitas timbangan boleh dianggap sama pada kedua skala neraca
yang berdekatan.
c) Repitabilitas digabungkan karena termasuk ke dalam presisi metode.
2) Ketidakpastian Baku Oven Diperoleh dari Kalibrasi Oven dan Efek Ruang
a) Kalibrasi Oven
Sertifikasi kalibrasi oven diperoleh data ± 6,18°C dengan tingkat
kepercayaan 95% dengan faktor cakupan dua. Evaluasi
ketidakpastiannya berdasarkan tipe B dengan asumsi distribusi normal.
Nilai ketidakpastian baku dari oven sebesar 3,09°C.
b) Ketidakpastian Efek Ruang
Ketidakpastian dari efek ruang dilakukan dengan percobaan kecil yaitu
contoh ditentukan kadar airnya pada suhu 105°C. Evaluasi
ketidakpastiannya berdasarkan tipe B. Nilai ketidakpastian baku dari efek
ruang oven sebesar 0,41009523%/°C. Hasil kalibrasi oven dan efek
ruang diperoleh ketidakpastian baku gabungannya adalah 1,267194%
c) Repitabilitas

35
Repitabilitas digabungakn karena termasuk ke dalam presisi metode.
Ketidakpastian baku kadar air (metode gravimetri) merupakan gabungan dari
komponen ketidakpastian baku di atas yaitu sebesar 1,268375%.
b. Ketidakpastian Baku Penimbangan (µc Mc)
Ketidakpastian baku penimbangan diperoleh 0,00411 gram dari kalibrasi,
sensitivitas, dan repitabilitas.
1) Kalibrasi Timbangan
Sertifikat kalibrasi timbangan yang diterbitkan oleh laboratorium kalibrasi yang
telah terakreditasi, tertulis perbedaan antara antara berat yang sebenarnya dengan
berat yang terbaca pada skala adalah ± 0,00582 gram dengan tingkat kepercayaan
95% dengan faktor cakupan dua. Evaluasi ketidakpastiannya berdasarkan tipe B
dengan asumsi distribusi normal. Nilai ketidakpastian kalibrasi harus dihitung dua
kali penimbangan contoh. Nilai ketidakpastian baku massa contoh yaitu 0,00114
gram.
2) Sensitivitas Timbangan
Sensitivitas timbangan diabaikan karena penimbangan berat dilakukan pada
timbangan yang sama dan perbedaan nilai yang kecil dari penimbangan wadah
sebelum dan sesudah ditambahkan contoh sehingga sensitivitas timbangan dapat
dianggap sama pada kedua skala neraca yang berdekatan.
3) Repitabilitas
Repitabilitas digabungkan karena termasuk ke dalam presisi metode.
c. Ketidakpastian Baku Volume Labu Takar (µc VLT)
Ketidakpastian baku volume labu akar diperoleh dari kalibrasi labu takar,
temperatur, dan repitabilitas. Hasil yang didapatkan sebesar 0,085020 mL.
1) Kalibrasi Labu Takar
Sertifikasi kalibrasi labu takar 100 mL grade A pabrikan Pyrex adalah ± 0,01 mL,
dengan tingkat kepercayaan 95% dengan faktor cakupan dua. Evaluasi
ketidakpastiannya berdasarkan tipe B dengan asumsi distribusi normal. Nilai
ketidakpastian bakunya adalah 0,005 mL.
2) Efek Temperatur
3) Temperatur laboratorium pada saat labu takar digunakan berbeda dengan
temperatur kalibrasi. Oleh karena itu, ketidakpastiannya efek temperatur harus

36
diperhitungkan. Menurut spesifikasi pabrik, labu takar dikalibrasi pada suhu 20°C.
Sedangkan variasi suhu di laboratorium ± 7°C. Ketidakpastian baku efek
temperatur dapat dihitung dari perbedaan antara temperatur laboratorium, volume
contoh, dan Koefisien Muai amonium asetat. Larutan amonium asetat 1 M
merupakan larutan encer sehingga untuk Koefisien Muai amonium asetat 1 M
dianggap sama dengan koefisien muai air (KMA) = 0,00021°C-1 dengan asumsi
distribusi segi empat berdasarkan evaluasi tipe B. Ketidakpastian baku efek
temperatur adalah 0,084873 mL.
4) Repitabilitas
Repitabilitas digabungkan termasuk ke dalam presisi metode. Hasil kalibrasi labu
takar dan efek diperoleh ketidakpastian baku gabungannya adalah 0,08502 mL.
d. Ketidakpastian Baku Recovery (µc Recovery)
Ketidakpastian baku Recovery harus diperhatikan untuk melihat apakah analit
terekstrak 100% dari matriks contoh. Untuk menghitung ketidakpastian Recovery dai
suatu metode diperlukan data Recovery yang dilakukan berulangkali dari suatu matriks
menggunakan meode tersebut. Nilai persentase Recovery diperoleh sebesar 103% dari
sepuluh kali pengulangan pengukuran sehingga diperoleh nilai Recovery sebesar
1,0375 dan nilai ketidakpastian bakunya diperoleh sebesar 0,06748. Adapun data
perhitungan ketidakpastian baku recovery dapat dilihat pada lampiran 7.
e. Ketidakpastian Baku Presisi Metode (µc PM)
Ketidakpastian presisi metode diperoleh dari pengukuran contoh sebanyak
sepuluh kali pengulangan, dihitung dalam bentuk Relative Standard Deviation (RSD).
Metode evaluasi ketidakpastiannya berdasarkan klasifikasi tipe A (metode statistika),
yaitu nilai standar deviasi yang diperoleh sama dengan nilai ketidakpastian bakunya.
Nilai ketidakpastian tersebut merupakan gabungan dari beberapa komponen
ketidakpastian dari repitabilitas, yaitu kadar air (metode gravimetri) penimbangan,
volume contoh, dan faktor pengenceran. Dalam penelitian ini diperoleh %RSD sebesar
4,2565% sehingga diperoleh nilai ketidakpastian baku presisi metode sebesar
0,042565. Adapun data perhitungan ketidakpastian baku presisi metode dapat dilihat
pada Tabel 19 lampiran 7.
Tabel 6. Nilai Ketidakpastian Baku Gabungan
Simbol Uraian Satuan Nilai (x) µ(x) µ(x)/(x)
KA Kadar Air % 43,06 1,268375 0,0294559

37
Mc Massa contoh gram 0,05 0,004110 0,0822000
VLT Volume Labu Takar mL 100 0,085020 0,0008502
Recovery Recovery - 1,0375 0,06748 0,0655194
PM Presisi Metode - - 0,042565 -
f. Ketidakpastian Diperluas (U)
Ketidakpastian diperluas merupakan hasil kali ketidakpastian gabungan dengan
faktor cakupan (k). Faktor cakupan (coverage factor) adalah faktor numerik yang
digunakan sebagai pangali terhadap ketidakpastian baku gabungan untuk memperoleh
ketidakpastian diperluas. Nilai k yang digunakan adalah 2 dengan selang kepercayaan
95%. Berdasarkan perhitungan ketidakpastian diperluasnya sebesar 1,4785%.
g. Pelaporan Hasil Uji
Penyajian pelaporan hasil uji dinyatakan dalam bentuk Y ± U. Y merupakan
nilai dari perhitungan kadar C-organik dalam pupuk organik dan U merupakan
ketidakpastiannya. Tingkat kepercayaan yang digunakan dalam pelaporan hasil uji
adalah 95% dengan faktor cakupannya 2. Pelaporan hasil uji yang diperoleh adalah
sebesar (6,7130 ± 1,4785)%. Rentang nilai ketidakpastiannya yang diperkirakan di
dalamnya terdapat nilai sebesar (8,1915 s/d 5,2345)%. Berdasarkan nilai
ketidakpastian yang diperoleh kontributor terbesar ketidakpercayaan penetapan kadar
C-organik dalam pupuk berasal dari recovery. Selain itu, kesalahan-kesalahan ini
dapat juga disebabkan oleh lingkungan. Untuk mendapatkan nilai ketidakpastian yang
kecil, sumber-sumber kesalahan tersebut harus diperkecil dengan lebih memperhatikan
efek-efek yang berpengaruh terhadapnya, yaitu efek suhu, kelembaban, getaran,
kompetensi, dari analis, homogenitas contoh, penggunaan instrumen, dan kalibrasi
dari peralatan yang digunakan. Menurut Komite Akreditasi Nasional (2003),
komponen yang lebih kecil dari satu per lima atau satu per tiga dari nilai
ketidakpastian baku nilainya tinggi dapat diabaikan.
BAB V
PENUTUP

38
A. Simpulan
Berdasarkan hasil validasi dan estimasi ketidakpastian metode penetapan C-organik
dalam pupuk organik menggunakan metode spektrofotometer diperoleh hasil sebagai
berikut:
1. Uji linieritas C-organik diperoleh persamaan regresi y = 0,0016x + 0,0037 dengan
nilai koefisien korelasi (r) sebesar = 0,9993.
2. Uji limit deteksi C-organik sebesar 59,8897 mg/L, jadi konsentrasi terkecil yang
dapat terbaca oleh alat instrumen adalah 59,8897 mg/L dan bila konsentrasi analit <
4,04 mg/L sinyal yang dihasilkan adalah noise.
3. Diperoleh kadar C-organik dalam sampel pupuk PO-02 BPTP Ungaran sebesar
7,6713%. Hasil kadar C-organik lebih dari 5% jadi pupuk organik koe PO-13 BPTP
Ungaran baik untuk digunakan sebagai pupuk tanaman yang dapat membuat lahan
menjadi produktif.
4. Uji presisi melalui repitabilitas C-organik dengan nilai persentase RSD sebesar
4,2565%.
5. Uji akurasi melalui persentase Recovery memperoleh nilai 104,44%.
6. Semua parameter validasi telah memenuhi syarat yang ditetapkan, jadi metode
penetapan C-organik dalam pupuk organik dengan metode spektrofotometer
dinyatakan valid sehingga dapat digunakan sebagai analisis rutin di laboratorium
tanah BPTP Jawa Tengah.
7. Estimasi ketidakpastian pada penetapan C-organik diperoleh persentase nilai
ketidakpastian pengukuran kadar air 1,268375; massa contoh 0,004110 gram;
volume labu takar 0,00085020 mL; Recovery 0,06748; dan presisi metode
0,042565.
8. Pelaporan hasil uji yang diperoleh adalah sebesar (6,7130 ± 1,4785)%. Rentang
nilai ketidakpastiannya yang diperkirakan di dalamnya terdapat nilai sebesar 8,1915
s/d 5,2345)%.
B. Saran
1. Praktikan hendaknya memahami terlebih dahulu tentang materi praktikum yang
akan dilakukan

39
2. Praktikan harus mengutamakan keselamatan kerja saat melakukan praktikum
seperti memakai jas laboratorium, sarung tangan, dan masker.
3. Sebisa mungkin saat pembacaan absorbansi C-organik dilakukan pada pagi hari,
karena pada siang hari kadar C-organiknya akan berubah.
4. Praktikan harus lebih teliti dan hati-hati dalam setiap langkah kerja.
5. Waktu yang digunakan saat pengovenan dan pendiaman larutan diusahakan sesuai
dengan yang ada di modul.
6. Perhatikan dengan cermat skala pada saat pengukuran.
DAFTAR PUSTAKA

40
Afandi, R.. 2002. Ilmu Kesuburan Tanah. Yogyakarta: Kanisus.
Ardana, Seta Kahardian. 2012. Validasi Metode Penetapan C-Organik Pada Pupuk Organik.
Laporan Prkatek Kerja Lapangan. Semarang: Kimia FMIPA Universitas Negeri
Semarang.
Balai Besar Litbang Sumberdaya Lahan Pertanian. 2010. Peranan Unsur Hara N, P, K dalam
Proses Metabolisme Tanaman Padi. Badan Penelitian dan Pengembangan Pertanian.
Bogor. 22 hal.
Basriman. 2011. Manfaat Pupuk Organik. http://distan.riau.go.id/index.php/
component/content/article/53-pupuk/149-manfaat-pupuk-organik. (Diakses pada
tanggal 20 Februari 2016).
Day, R. A. dan A. L. Underwood. 1992. Analisis Kimia Kuantitatif. Diterjemahkan oleh
Aloysios Hadyana Pudjaatmaka. Erlangga. Jakarta.
Fauzi, Ahmad. 2008. Analisis Kadar Unsur Hara Karbon Organik dan Nitrogen di Dalam
Tanah Perkebunan Kelapa Sawit Bengkalis Riau. Medan: Univesitas Sumatera Utara
Press.
Hardjowigeno. 2004. Klasifikasi Tanah dan Pedogenesis. Jakarta: Akademia Pressido
Hanolo.
Harmita. 2004. Petunjuk Pelaksanaan Validasi Metode dan Cara Pelaksanaannya. Jakarta:
Majalah Ilmu Kefarmasian, Vol.1. Departemen Farmasi FMIPA-UI.
Hendayana, Sumar dkk.. 1994. Kimia Analitik Instrumen. Semarang: IKIP Semarang Press.
Komite Akreditasi Nasional. 2003. Pedoman Evaluasi dan Pelaporan Ketidakpastian
Pengukuran. Komite Akreditasi Nasional. Jakarta.
Lingga, P. dan Marsono. 2000. Petunjuk Penggunaan Pupuk. Jakarta: Penebar Swadaya.
Miller, J.C. dan J.N. Miller. 1991. Statistika untuk Kimia Analitik. Edisi Kedua.
Diterjemahkan oleh Drs. Suroso, M.Sc. Bandung: ITB.
Natalia, Cisca. 2009. Akurasi dan Presisi. http://hardipurba.com/2009/06/30/akurasi-dan-
presisi.html . (diakses pada tanggal 20 Februari 2016).
SNI 19-17025-2000. 2000. Persyaratan Umum Kompetensi Laboratorium Penguji dan
Laboratorium Kalibrasi. Badan Standarisasai Nasional. Jakarta.
Eviati dan Sulaeman. 2012. Petunjuk Teknis Analisis Kimia Tanah, Tanaman, Air dan Pupuk.
Bogor: Balai Penelitian Tanah Badan Penelitian dan Pengembangan Pertanian.
Sumardi. 2002. Validasi Metode Pengujian. Bogor: Pusat Standarisasi dan Akreditasi
Seketariat Jenderal Departemen Pertanian.
Wulandari, Indah. 2007. Ilmu Tanah. Jepara: Nusa Perdana.

41
Lampiran 1
1. Diagram Alir Penetapaan Kadar Air (Metode Gravimetri)

Mulai
Sampel Pupuk Organik Kode
PO-02
Cawan Porselin (Diketahui beratnya)
5,000 gram sampel dimasukkan ke dalam cawan
Dioven pada suhu 105°C selama 3 jam
Cawan+sampel diangkat dengan penjepit dan
dimasukkan desikator 15 menit
Ditimbang dengan neraca analitik
Tabel Data Pengamatan Kadar
Air
Selesai
42
Gambar 5. Diagram Alir Penetapaan Kadar Air (Metode Gravimetri)
2. Diagram Alir Pembuatan Larutan Standar C-Organik

Mulai
Larutan Standar baku 5000 mg/L
Larutan K2Cr2O7 1 N
H2SO4 pa 98%
Dipipet Larutan standar 0 mL, 1 mL, 2 mL, 3 mL, 4 mL, dan 5 mL masing-masing untuk 0 ppm, 50 ppm, 100 ppm, 150 ppm, 200 ppm, dan 250
dalam labu takar 100 mL ppm
Masing-masing ditambahkan 5 mL larutan Larutan K2Cr2O7 1 N,
dikocok
Aquades
Masing-masing ditambahkan 7,5 mL H2SO4 pa 98%, dikocok
Didiamkan sampai dingin
Ditambah aquades sampai tanda batas
Selesai
43
Gambar 6. Diagram Alir Pembuatan Larutan Standar C-Organik

Mulai
Sampel Pupuk Organik 0,05 gram
Larutan K2Cr2O7 1 N
H2SO4 pa 98%
Sampel dimasukkan ke dalam labu takar 100 mL
Masing-masing ditambahkan 5 mL larutan Larutan K2Cr2O7 1 N,
dikocok
Aquades
Masing-masing ditambahkan 7,5 mL H2SO4 pa 98%, dikocok
Didiamkan sampai dingin
Ditambah aquades sampai tanda batas
Selesai
44
3. Diagram Alir Pembuatan Larutan Sampel C-Organik
Gambar 7. Diagram Alir Pembuatan Larutan Sampel C-Organik

Mulai
Dipipet 10 mL standar baku C ke dalam labu takar 50 mL
Larutan Standar Baku C 5000 ppm Aquades
Dihipitkan larutan sampai tanda batas dengan aquades
Selesai
45
4. Diagram Alir Pembuatan Larutan Spike 1000 mg/L
Gambar 8. Diagram Alir Pembuatan Larutan Spike 1000 mg/L

Mulai
Sampel Pupuk Organik 0,05 gram
Larutan K2Cr2O7 1 N
H2SO4 pa 98%
Sampel dimasukkan ke dalam labu takar 100 mL
Masing-masing ditambahkan 5 mL larutan Larutan K2Cr2O7 1 N,
dikocok
Aquades
Masing-masing ditambahkan 7,5 mL H2SO4 pa 98%, dikocok
Didiamkan sampai dingin
Ditambah aquades sampai tanda batas
Selesai
Ditambahkan larutan spike 2 mL
Larutan Spike
46
5. Diagram Alir Pembuatan Larutan Contoh + Spike
Gambar 9. Diagram Alir Pembuatan Larutan Contoh + Spike

47
Lampiran 2
Data Linieritas C-Organik
Tanggal pembuatan deret standart : 10 Februari 2016
Tanggal Pengukuran : 11 februari 2016
Tabel 7. Linearitas C-Organik
UlanganAbsorbansi Standart
slope R2
0 ppm 50 ppm 100 ppm 150 ppm 200 ppm 250 ppm
1 0 0,085 0,162 0,245 0,318 0,391 0,0016 0,9992
2 0 0,073 0,158 0,238 0,316 0,391 0,0016 0,9996
3 0 0,080 0,160 0,241 0,307 0,390 0,0015 0,9993
4 0 0,084 0,162 0,241 0,320 0,387 0,0016 0.9991
5 0 0,085 0,162 0,244 0,326 0,393 0,0016 0,9991
6 0 0,085 0,168 0,248 0,323 0,391 0,0016 0,9984
7 0 0,085 0,165 0,245 0,323 0,390 0,0016 0,9988
8 0 0,087 0,165 0,245 0,323 0,387 0,0016 0,9983
9 0 0,076 0,154 0,238 0,316 0,383 0,0016 0.9992
10 0 0,082 0,160 0,241 0,307 0,390 0,0016 0,9992
x 0 0,082 0,162 0,243 0,318 0,389 0,0016 0,9993
Gambar 10. Grafik Linearitas
C-organik
0 50 100 150 200 250 3000
0.050.1
0.150.2
0.250.3
0.350.4
0.45
f(x) = 0.00156228571428571 x + 0.0037142857142857R² = 0.999309618284688
Kurva Kalibrasi Standar C-Organik
Konsentrasi (ppm)
Abso
rban
si
Konsentrasi
standar (ppm)
Absorbansi
0 0
50 0,082
100 0,162
150 0,243
200 0,318
250 0,389

48
Perhitungan linearitas standar C-organik,
Persamaan regresi: y = a + bx
a = [∑ Y −(b∑ X )]/ n
b = ∑ XY −[ (∑ XY ) /n ]∑ x2−[∑ x2 /n ]
r = ∑ XY −[(∑ XY )/n ]
∑ x2−(∑ x2)
n √∑ y2−(∑ y2 )
n
Hasil dari perhitugan dari kurva kalibrasi:
y = 0,0037 + 0,0016x
slope = 0,0016
intersep = 0,0037
r = 0,9993

49
Lampiran 3
Data dan Contoh Perhitungan Limit Deteksi C-Organik
Tanggal pembentukan ekstrak : 10 Februari 2016
Tanggal pengukuran : 11 Februari 2016
Contoh : Pupuk Organik Kode PO-2
Intersep : 0,0037
Slope : 0,0016
Tabel 8. Data Perhitungan Limit Deteksi
Ulangan
Absorbansi Konsentrasi (ppm) Konsentrasi
Standar C-Organik
(ppm)BlankoStandar
TerendahBlanko
Standar
Terendah
1 0 0,085 -2,3125 50,8125 53,125
2 0 0,073 -2,3125 43,3125 45,625
3 0 0,080 -2,3125 47,6875 50,000
4 0 0,084 -2,3125 50,1875 52,500
5 0 0,085 -2,3125 50,8125 53,125
6 0 0,085 -2,3125 50,8125 53,125
7 0 0,085 -2,3125 50,8125 53,125
8 0 0,087 -2,3125 52,0625 54,375
9 0 0,076 -2,3125 45,1875 47,500
10 0 0,082 -2,3125 48,9375 51,250
N 10
x 51,375
SD 2,8382
LOD 8,5147
LD 59,8897

50
Contoh perhitungan:
Ulangan 1
Konsentrasi yang diperoleh:
Konsentrasi Blanko
y = 0,0016x + 0,0037
0 = 0,0016x + 0,0037
-0,0037 = 0,0016x
x = -2,3125
Konsentrasi Standar Terendah
y = 0,0016x + 0,0037
0,085 = 0,0016x + 0,0037
0,085 - 0,0037 = 0,0016x
0,0813 = 0,0016x
x = 50,8125 mg/L
Konsentrasi rata-rata = (53,125 + 45,625 + 50,000 + 52,500 + 53,125 + 53,125
+ 53,125 + 54,375 + 47,500 + 51,250) mg/L
7 = 51,375 mg/L
SD = 2,8382
LOD = 3SD
= 8,5147
Limit Deteksi Instrumen = Nilai
rata-rata konsentrasi terkecil + 3SD
= 51,375 mg/L + 8,5147 mg/L

51
= 59,8897 mg/L
Lampiran 4
Data dan Contoh Perhitungan Repitabilitas C-Organik
Tanggal pembuatan ekstrak : 10 Februari 2016
Tanggal pengukuran : 11 Februari 2016
Tabel 9. Data repitabilitas C-Organik
UlanganMassa Contoh
(mg)
Absorbansi
Contoh (Abs)
Konsentrasi
Contoh (ppm)C-Organik (%)
1 50,4 0,036 20,1875 7,0344
2 50,9 0,034 18,9375 6,5339
3 50,1 0,032 17,6875 6,2002
4 50,1 0,034 18,9375 6,6383
5 50,1 0,039 22,0625 7,7338
6 50,5 0,034 18,9375 6,5857
7 50,1 0,035 19,5625 6,8574
8 50,6 0,033 18,3125 6,3558
9 50,2 0,034 18,9375 6,6251
10 50,3 0,036 20,1875 7,0484
N 10
x 6,7613
SD 0,2878
%RSD 4,2565
Contoh Perhitungan
Ulangan 1:
Konsentrasi ekstrak
y = bx + a
y = 0,0016x + 0,0037
0,033 = 0,0016x + 0,0037

52
x = 18,3125 mg/L
Kadar C-Organik (%) = ppm kurva x (100/mg contoh) x (100 mL/1000 mL) x fk
= 20,1875 x (100/50,4) x (0,1) x 1,7562
= 7,0344%
SD = √∑ (xi−x )2
n−1
SD = 0,2878
% RSD = SDx x 100%
= 0,28786,7613 x 100%
= 4,2565%

53
Lampiran 5
Data dan Contoh Perhitungan Recovery C-Organik
Tanggal pembuatan ekstrak : 10 Februari 2016
Tanggal pengukuran : 11 Februari 2016
Contoh : Pupuk Organik PO-2
Berat contoh : 0,05 gram
Spike yang ditambahkan : 2 mL 1000 ppm C
Berat spike sebagai C : 2 mg C
Data dari linearitas : slope = 0,0016 dan intersep = 0,0037
Tabel 10. Hasil Perhitungan Recovey C-Organik (%)
No.
Absorbansi Kadar C-Organik%
RecoveryConto
h
Contoh
+Spike
Contoh Contoh + Spike
mg ppm % mg ppm %
10,036
0,07250,4 20,1875
7,034
4
50,3 42,687514,9041
112,50
20,034
0,06550,9 18,9375
6,533
9
50,3 38,312513,3766
96,87
30,032
0,07050,1 17,6875
6,200
2
50,1 41,437514,5254
118,75
40,034
0,06450,1 18,9375
6,638
3
50,1 37,687513,2109
93,75
50,039
0,06550,1 22,0625
7,733
8
50,1 38,312513,4300
81,25
60,034
0,06950,5 18,9375
6,585
7
50,2 40,812514,2779
109,37
70,035
0,06950,1 19,5625
6,857
4
50,8 40,812514,1092
106,25
8 0,033 0.065 50,6 18,3125 6,355 50,7 38,3125 13,2711 100,00

54
8
90,034
0,06850,2 18,9375
6,625
1
50,9 40,187513,8659
106,25
100,036
0,07250,3 20,1875
7,048
4
50,6 42,687514,8158
112,50
x 0,0347 0,0679 50,3 19,3750 6,761
3
50,4 40,1250 13,9816 103,75
Contoh perhitungan
x C3 = CBFp
= 1000
50
= 20 mg/L
Ulangan 1
Konsentrasi Contoh
y = bx + a
0,036 = 0,0016x + 0,0037
x = 0,03230,0016
x = 20,1875 mg/L .......................................................... (C1)
Kadar C-Organik (%) Contoh + spike = ppm kurva x (100/mg contoh) x (100
mL/1000 mL) x fk
= 42,6875 x (100/50,3) x 0,1 x 1,7562
= 14,9041%
Konsentrasi Contoh + Spike
y = bx + a

55
0,088 = 0,0016x + 0,0037
x = 0,08430,0016
x = 52,6875 mg/L .......................................................... (C2)
Konsentrasi Spike = 20 mg/L ........................................... (C3)
Perhitungan Akurasi
% Recovery = C 2−C 1
C 3 x 100%
= 42,6875−20,1875
20 x 100%
= 112,5%
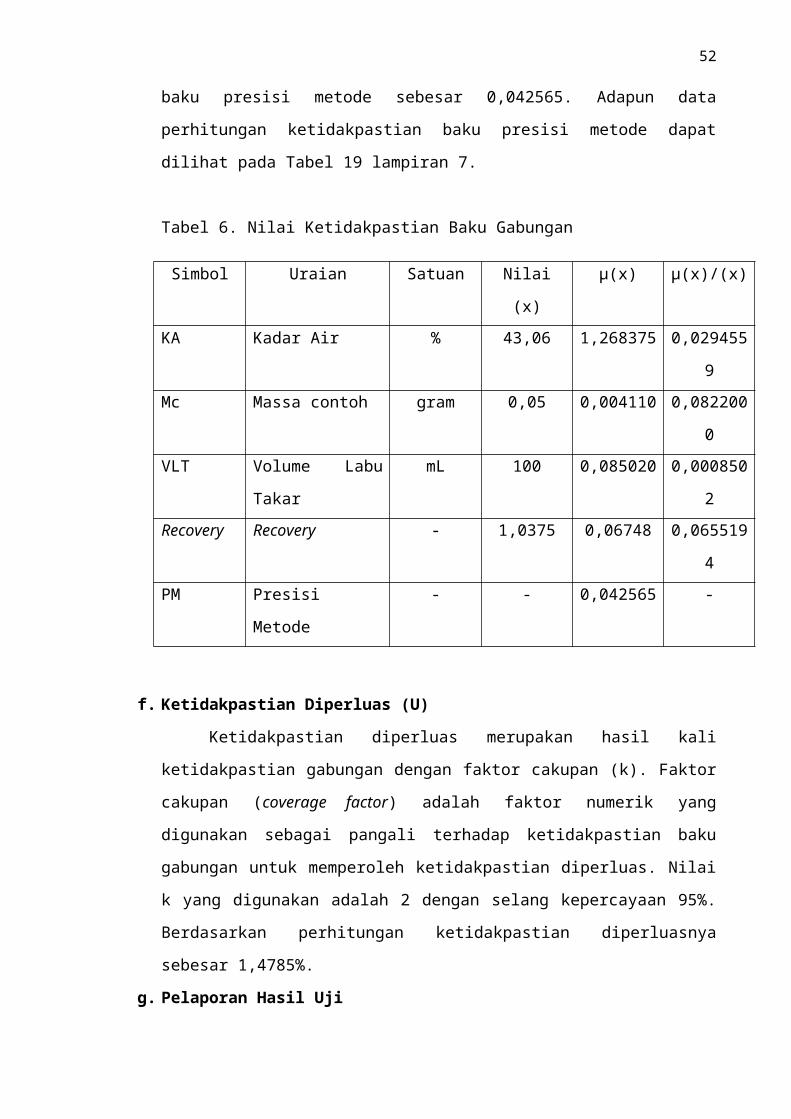
56
Lampiran 6
Penentuan Kadar Air dan Faktor Koreksi BKM
Tanggal Penetapan : 11 Februari 2016
Sampel : Pupuk Organik PO-2
Berat contoh : 10 gram
Tabel 11. Penetapan Kadar Air dan Faktor Koreksi BKM
No
Berat
cawan
kosong
(M1)
Berat
cawan+s
ampel
(M2)
Berat
setelah
dioven
(M3)
Bobot
sampel
Bobot yang
hilang
Kadar
Air
(KA)%
Faktor
koreksi
BKM
1 22,41 32,41 28,14 10 4,27 42,7 1,7452
2 21,99 31,99 27,69 10 4,30 43,0 1,7544
3 21,12 31,12 26,81 10 4,31 43,1 1,7575
4 21,72 31,72 27,50 10 4.22 42,2 1,7301
5 21,93 31,93 27,62 10 4,31 43,1 1,7575
6 21,02 31,02 26,70 10 4,32 43,2 1,7606
7 22,09 32,09 27,83 10 4,26 42,6 1,7422

57
8 22,10 32,10 27,71 10 4,39 43,9 1,7825
9 21,38 31,38 27,02 10 4,36 43,6 1,7730
10 22,27 32,27 27,95 10 4,32 43,2 1,7606
x 21,803 31,803 27,497 10 4,306 43,06 1,7562
Perhitungan Kadar Air
KA (%) = (M2-M3)(M2-M1) x 100%
= (31,803 -27,497)(31,803-21,803) x 100%
= 4,306
10 x 100%
= 43,06%
Perhitungan faktor koreksi BKM
fk = 100100-%kadar air
= 100100-43,06
= 1,7562

58
Lampiran 7
Data dan Perhitungan Estimasi Ketidakpastian Penetapan C-Organik dalam Contoh
Pupuk Organik
1. Formula atau persamaaan
Kadar C-Organik (%) = ppm kurva x (100/mg contoh) x (100 mL/1000 mL) x fk
= 20,1875 x (100/50,4) x (0,1) x 1,7562
= 20,1875 x 1,9841 x 0,1 x 1,7562
= 7,0344%
Keterangan:
ppm kurva : kadar contoh yang didapat dari kurva hubungan antara
kadar deret standar dengan pembacaannya setelah di-
koreksi blanko
fk : faktor koreksi kadar air – {100/(100-%kadar air)}
2. Daftar sumber ketidakpastian
Tabel 12. Sumber-sumber ketidakpastian
Simbol Uraian Nilai Satuan
KA Kadar Air 43,06 %

Recovery Mc
Kal
KA VLT
Kal
ET
Karbon Organik
Kurva KalibrasiPM
Oven
Massa C3
59
Mc Massa contoh 0,05 Gram
VLT Volume Labu Takar 100 Gram
Recovery Recovery 1,0375 -
PM Presisi Metode - -
3. Cause and effect diagram atau fish bone
Gambar 3. Cause and effect diagram atau fish bone C-organik
4. Sumber-sumber estimasi ketidakpastian
a. Ketidakpastian Baku Kadar Air (Metode Gravimetri) (µc KA)
1) Formula atau persamaan
KA (%) = (M2-M3)(M2-M1) x 100%

Massa Teruapkan
Kadar Air(%)
Massa Basah Oven
M2
M2
Ka
Ka
M3
Ka
KaM1 Ka
ET
60
= (31,803-27,497)(31,803-21,803) x 100%
= 4,306
10 x 100%
= 43,06%
2) Daftar Data Sumber Ketidakpastian
Tabel 13. Sumber ketidakpastian kadar air
Simbol Uraian Nilai Satuan
M1 Bobot Kosong 21,803 Gram
M2 Bobot Kosong + Contoh
Sebelum Dipanaskan
31,803 Gram
M3 Bobot Kosong + Contoh
Setelah Dipanaskan
27,497 Gram
KA Kadar Air 43,06 %
3) Cause and effect diagram atau fish bone
Gambar 4. Cause and effect diagram atau fish bone Kadar Air Sampel
Keterangan:

61
Ka : Kalibrasi
M1 : Bobot Kosong
M2 : Bobot Kosong + Contoh Sebelum Dipanaskan
M3 : Bobot Kosong + Contoh Setelah Dipanaskan
ET : Efek Temperatur
4) Sumber Estimasi Ketidakpastian Baku (µc)
a) Ketidakpastian Baku Massa Teruapkan (µc M2-M3)
Sertifikasi kalibrasi neraca ± 0,00582 gram, pada tingkat kepercayaan
95%.
Evaluasi tipe B, k=2
µc Kal = Qk =
0,00582 ggram2 = 0,00291 gram
b) Ketidakpastian Baku Massa Basah (µc M2-M1)
Sertifikasi kalibrasi neraca ± 0,00582 gram, pada tingkat kepercayaan
95%.
Evaluasi tipe B, k=2
µ Kal = Qk =
0,00582 gram2 = 0,00291 gram
µc Mc = √2 x (µ Kal )2 = √2 x (0,00291 )2 = 0.00411 gram
c) Ketidakpastian Baku Efek Suhu Oven (µc efek suhu/ruang)
Sertifikasi kalibrasi ± 6,18°C pada tingkat kepercayaan 95%.
Evaluasi tipe UM 400/E496.0679, k=2
µ Kal = Qk =
6,18° C2 = 3,09°C
Evaluasi tipe B
µc efek ruang = KA %
Suhu °C = 43,06105 = 0,41009523%/°C
Ketidakpastian baku efek ruang
µ (efek/ruang) = µ Kal x µ efek ruang
= 3,09°C x 0,41009523%/°C
= 1,267194%
5) Ringkasan Nilai Ketidakpastian Kadar Air
Tabel 14. Ringkasan Ketidakpastian Kadar Air
Sumber Satuan Nilai (x) µ(x) µ(x)/(x)

62
Massa Teruapkan Gram 4,306 0.00411 0,000954
Massa Basah Gram 10 0.00411 0,000411
Oven % 43,06 1,267194 0,029428
6) Ketidakpastian Gabungan dari Kadar Air
µc (KA) = KA% x
√( µ Massa teruapkanMassateruapkan
)2
+( µ Massa basahMassa basah
)2
+( µOvenOven
)2
= 43,06% x √(0,000954 )2+(0,000411)2+(0,029428)2
= 43,06% x √(0,910)−7+(0,168)−7+(0,86659 )−4
= 43,06% x √0,0008676738
= 43,06 x 0,029456
= 1,268375%
b. Ketidakpastian Baku Massa Contoh (µc Mc)
Sertifikasi kalibrasi neraca ± 0,00582 gram, pada tingkat kepercayaan 95%.
Evaluasi tipe B, k=2
µ Kal = Qk =
0,00582 gram2 = 0,00291 gram
µc Mc = √2 x (µ Kal )2 = √2 x (0,00291 )2 = 0.00411 gram
c. Ketidakpastian Baku Volume Labu Takar (µc VLT)
Ketidakpastian labu takar 100 mL
1) Sertifikasi kalibrasi ± 0,01 mL pada tingkat kepercayaan 95%
Evaluasi tipe B, k=2
µ Kal = Qk =
0,01 mL2 = 0,005 mL
2) Efek Temperatur (ET)
Evaluasi tipe B, k = √3Variasi suhu = 27°C-20°C = 7°C
Koefisien Muai Air = 0,00021°C-1
µ ET = Variasi xVLT xkoefisien muai air
k
= 7 °C x 100 mL x0,00021 °C−1
√3

63
= 0,147
1,7320
= 0,084873 mL
Ketidakpastian baku gabungan dari volume labu takar
µ (VLT) = √(µKal)2+(µET )2
= √(0,005)2+(0,084873)2
= √0,000025+0,007203
= √0,007228
= 0,08502 mL
d. Ketidakpastian Baku Recovery (µc Recovery)
Tabel 15. Data Hasil Ringkasan Recovery
Ulangan
Konsentrasi
Contoh (mg/L)
Konsentrasi Spike
(mg/L)
Konsentrasi Contoh+Spike
(mg/L)
C1 C3 C2
1 20,1875 20,00 42,6875
2 18,9375 38,3125
3 17,6875 41,4375
4 18,9375 37,6875
5 22,0625 38,3125
6 18,9375 40,8125
7 19,5625 40,8125
8 18,3125 38,3125
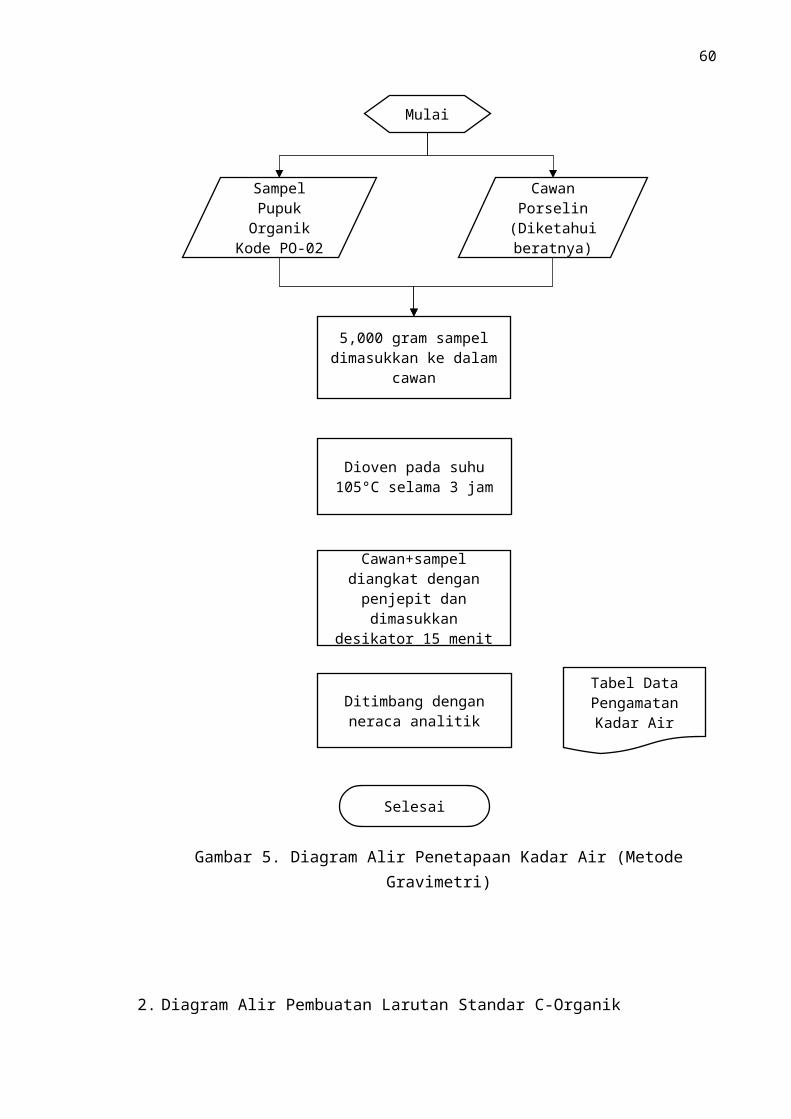
64
9 18,9375 40,1875
10 20,1875 42,6875
x 19,3750 40,1250
SD 1,21630 1,87270
(SD)2 1,47930 3,50700
Recovery = C 2−C 1
C 3
= 50,2500−19,3750
29,5625
= 30,875
29,5625
= 1,044
Ketidakpastiaan baku konsentrasi spike larutan baku (μ xC 3¿
1) Formula dan persamaan
xC 3=CBFp
=¿ 100050 = 20 mg/L Fp= VLT
V std=100 ml
2 ml = 50
Keterangan :
V std : Volume standar spike yang ditambahkan (mL)
VLT : Volume Labu Takar (mL)
2) Daftar Data Sumber Ketidakpastian
Tabel 16. Daftar Data Sumber Ketidakpastian x C3
Simbol Uraian Nilai Satuan
CB Konsentrasi Baku 1000 mg/L
Fp Faktor Pengenceran 50 -
x C 3 Rata-rata konsentrasi spike larutan baku 29,5625 mg/L
3) Cause and Effect Diagram atau Fish Bone

65
Gambar 11. Cause and Effect Diagram atau Fish Bone x C3
Keterangan :
ET : Efek Temperatur
Kal : Kalibrasi
Rep : Repitabilitas
4) Sumber Estimasi Ketidakpastian Baku
Ketidakpastiaan Baku Faktor Pengenceran (Fp) dan Konsentrasi Baku (CB)
a) Ketidakpastiaan Baku Volume Standar (μc V stdr ¿
i. Ketidakpastian baku pipet ukur 2 ml
Sertifikat kalibrasi ± 0,0029 mLpada tingkat kepercayaan 95 %
Evaluasi tipe B, k = 2
μ Kal=Quk
=0,00292
=0,00145 ml
ii. Efek Temperatur (ET)
Evaluasi tipe B, k = √3
Variasi suhu = 7℃Koefisien Muai Air (KMA) = 0,00021 ℃-1
μ ET=Variasi suhu xV std X KMAk
μ ET=7℃ x2ml x 0,00021℃−1
√3
μ ET=0,001699mL
iii. Repitabilitas (Rep)

66
Standar deviasi pengukuran volume 2 ml = 0,00878 ml
μ Rep=0,00878 ml
Ketidakpastian baku gabungan dari volume labu takar 1
μc (V stdr )=√¿¿
μc (V stdr )=√¿¿
μc (V stdr )=0,0090546 mL
b) Ketidakpastiaan Baku Volume Labu Takar (μcVLT ¿
Ketidakpastian baku labu takar 100 ml
i. Sertifikat kalibrasi ± 0,01 mlpada tingkat kepercayaan 95 %
Evaluasi tipe B, k = 2
μ Kal=Quk
=0,012
=0,005 ml
ii. Efek Temperatur (ET)
Evaluasi tipe B, k = √3
Variasi suhu = 7℃Koefisien Muai Air (KMA) = 0,00021 ℃-1
μ ET=Variasi suhu xVLT X KMAk
μ ET=7℃ x100 ml x 0,00021℃−1
√3
μ ET=0,0848729m l
iii. Repitabilitas (Rep)
Standar deviasi pengukuran volume 100 ml = 0,0234 ml
μ Rep=0,0234 ml
Ketidakpastian baku gabungan dari volume labu takar 1
μc (VLT )=√¿¿
μc (VLT )=√¿¿
μc (VLT )=0,0881476 mL
Ketidakpastian baku gabungan faktor pengenceran
μc ( Fp )=Fp x √( μ V stdV std )
2
+( μVLTVLT )
2
= 50 x√( 0,0090546 ml2ml )
2
+( 0,0881476 ml100 ml )
2

67
= 50 x 0,004611
= 0,23058
Ketidakpastian Gabungan Konsentrasi Baku
μc (CB )=1000 mgL
x √¿¿
μc (CB )=1000 mgL
x √¿¿
μc (CB )=1000 mgL
x √0,000000777+0,00002126
μc (CB ) = 1000 mgL x 0,004694
μc (CB )=4,694 mg / L
5) Ringkasan dari Nilai Ketidakpastian Konsentrasi Spike Larutan Baku
Tabel 17. Ringkasan Nilai Ketidakpastian Konsentrasi Spike Larutan Baku
Simbol Uraian Satuan Nilai (x) μ(x ) μ(x )/(x)
CB Konsentrasi baku mg/L 1000 4,694 0,004694
Fp Faktor Pengenceran - 50 0,23058 0,004612
6) Ketidakpastiaan gabungan konsentrasi spike larutan baku
μc ( x C 3 )=Konsentrasi Spike x√( μCBCB )
2
+( μ FpFp )
2
= 20 mg/L x √(0,004694 )2+(0,004612)2
= 20 mg/L x 0,0065215
= 0,13043 mg/L
µ Recovery = Recovery x √ SD2C 2n
+SD 2C 1
(C 2−C 1)2 +( µC 3C 3
)2
= 1,03 x √ 3,50710
+1,4793
(20,75)2 +( 0,1304320
)2
= 1,03 x √ 0,3507+1,4793430,5625
+0,00004253
= 1,03 x √0,0042503+0,00004253

68
= 1,03 x √0,0042928
= 1,03 x 0,0655
= 0,06748
e. Ketidakpastian Baku Presisi Metode (µc PM)
Tabel 18. Data Hasil Presisi Metode
Ulangan Bobot
Contoh
(mg)
Absorbans
i Contoh
(Abs)
Absorbansi
Blanko
(Abs)
Konsentras
i (mg/L) C-Organik (%)
1 50,4 0,036 0 20,1875 7,0344
2 50,9 0,034 0 18,9375 6,5339
3 50,1 0,032 0 17,6875 6,2002
4 50,1 0,034 0 18,9375 6,6383
5 50,1 0,039 0 22,0625 7,7338
6 50,5 0,034 0 18,9375 6,5857
7 50,1 0,035 0 19,5625 6,8574
8 50,6 0,033 0 18,3125 6,3558
9 50,2 0,034 0 18,9375 6,6251
10 50,3 0,036 0 20,1875 7,0484
x 50,3 0,0347 0 19,3750 6,7613
SD 0,2878
%RSD 4,2565
Evaluasi tipe A
µc (PM) = RSD = %RSD
100 = 0,042565
f. Nilai Ketidakpastian Baku Gabungan
Tabel 6. Nilai Ketidakpastian Gabungan
Simbol Uraian Satuan Nilai (x) µ(x) µ(x)/(x)
KA Kadar Air % 43,06 1,268375 0,0294559
Mc Massa contoh gram 0,05 0,004110 0,0822000

69
VLT Volume Labu Takar mL 100 0,085020 0,0008502
Recovery Recovery - 1,0375 0,06748 0,0655194
PM Presisi Metode - - 0,042565 -
g. Ketidakpastian baku gabungan penetapan C-Organik dalam contoh pupuk
secara Spektrofotometer (tanpa memperhitungkan presisi mode)
y = 6,7613%
µ (C-Organik) = y x √( µKAKA
)2
+( µMcMc
)2
+( µVLTVLT
)2
+( µRecoveryRecovery
)2
= 6,7613% x
√(0,0294559)2+(0,0822)2+(0,0008502)2+(0,0655194 )2
= 6,7613% x √0,0119172
= 6,7613% x 0,109165
= 0,7381%
h. ketidakpastian baku gabungan penetapan C-organik dalam contoh pupuk secara
Spektrofotometer (dengan memperhitungkan presisi mode)
µc (C-Organik) = √(µC−Organik)2+(µ PM )2
= √ (0,7381 )2+ (0,042565 )2
= √0,5466
= 0,7393%
i. Ketidakpastian diperluas (U)
Pada tingkat kepercayaan 95% k=1
U = µc (C-Organik) mg/L x 2
U = 0,7393% x 2
U = 1,4785%
j. Pelaporan hasil uji
Pada tingkat kepercayaan 95%
(Y ± U)%
(6,7130 ± 1,4785)%
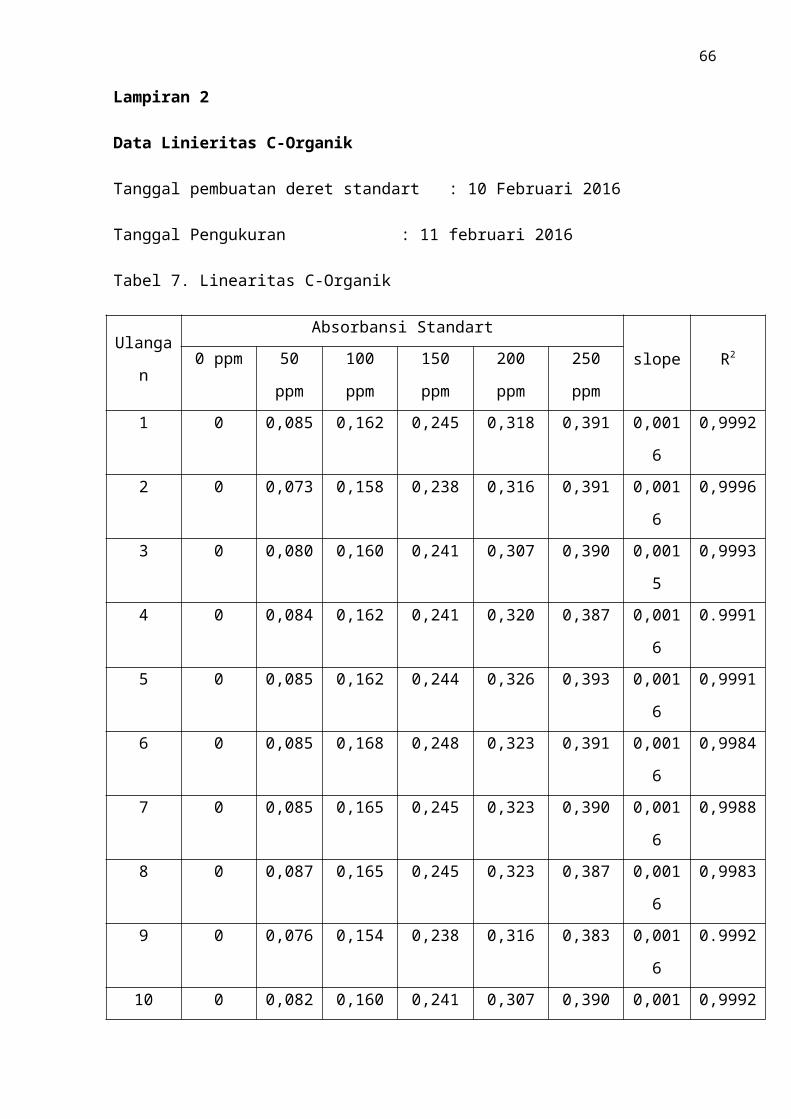
70
Lampiran 8
DOKUMENTASI
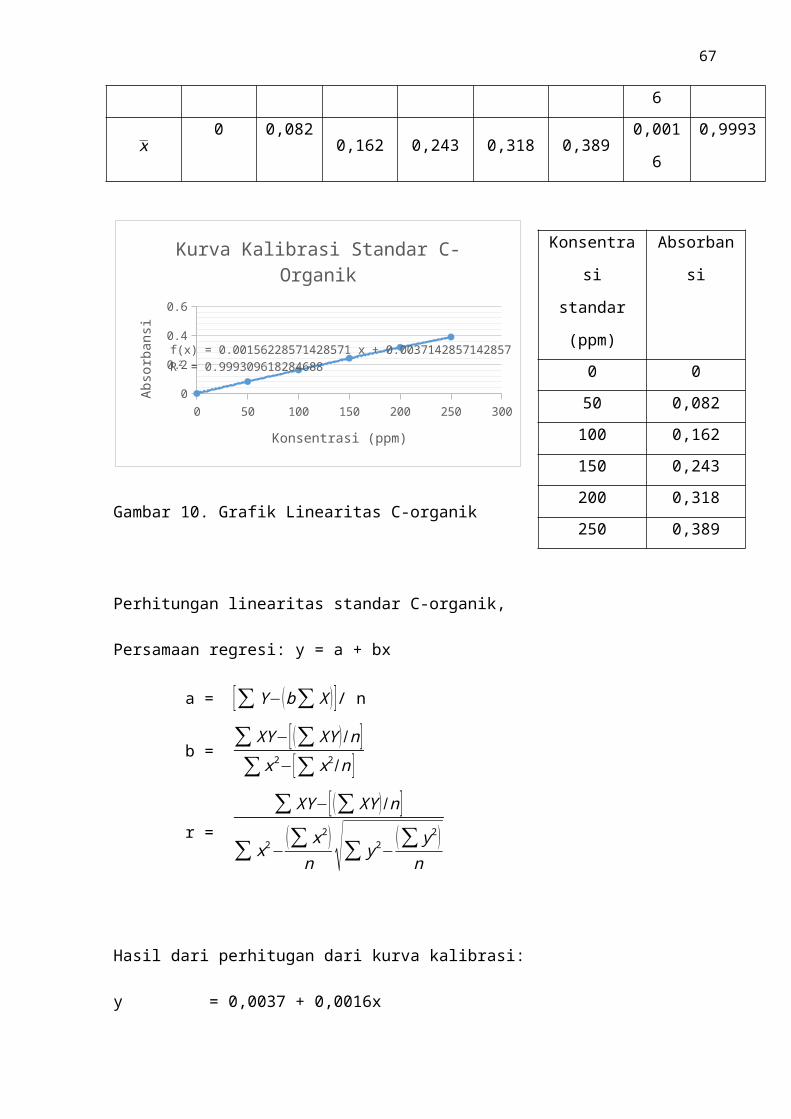
71
Kantor BPTP Jawa Tengah Laboratorium Kimia BPTP Jawa Tengah
Sampel Pupuk Organik PO-02 Deksikator

72
Penimbangan Sampel Kadar Air Pupuk Organik
Pengovenan Sampel Kadar Air Pupuk Organik
Penimbangan Sampel Pupuk Organik Untuk Larutan
Penambahan K2Cr2O7 Pada Larutan Standar dan Sampel Pupuk Organik
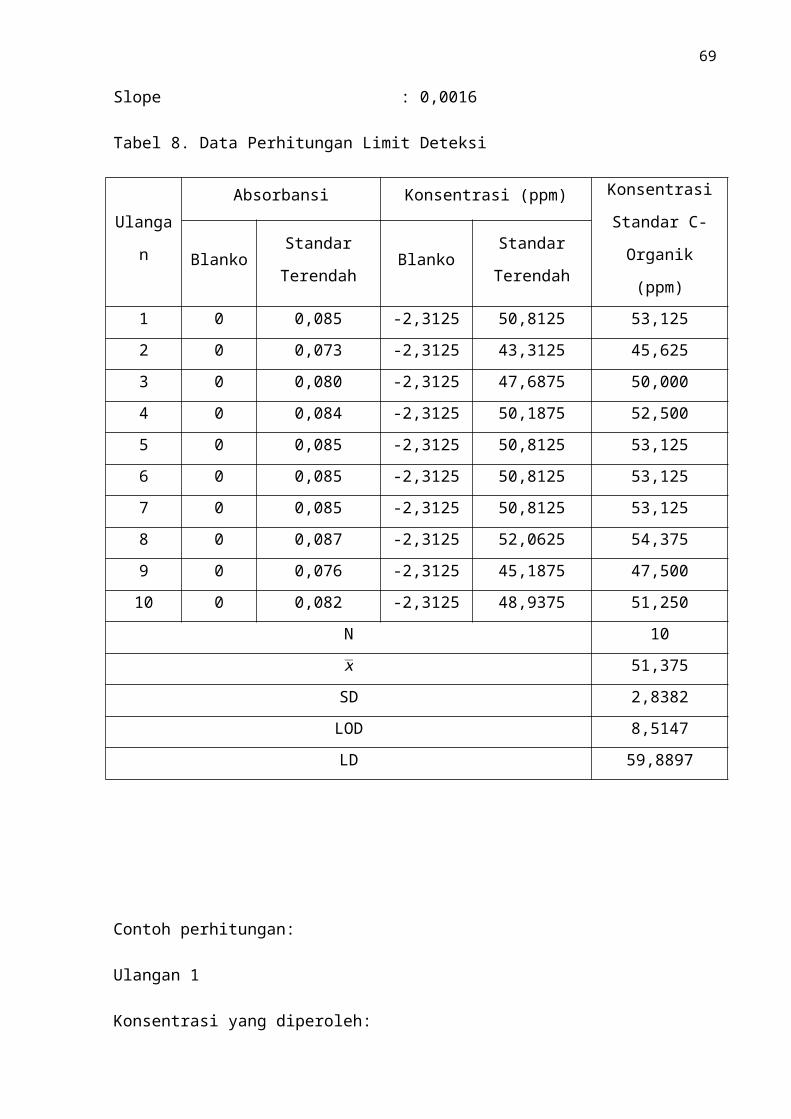
73
Penambahan H2SO4
Pengenceran Larutan Standar dan Larutan Sampel Pupuk Organik
Larutan Standar 250 ppm, 200 ppm, 150 ppm, 100 ppm, 50 ppm, dan 0 ppm
Larutan Sampel Pupuk Organik
Kuvet Berisi Larutan Standar Pembacaan Absorbansi dengan Spektrofotometer